
More Information
Submitted: February 08, 2021 | Approved: March 03, 2021 | Published: March 04, 2021
How to cite this article: Madkour LH. Imaging modalities delivery of RNAi therapeutics in cancer therapy and clinical applications. J Radiol Oncol. 2021; 5: 005-034.
DOI: 10.29328/journal.jro.1001035
Copyright License: © 2021 Madkour LH. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Molecular imaging; Cancer; Nanocarrier; siRNA; RNA interference; Gene delivery; Theranostics; X-ray computed tomography

Imaging modalities delivery of RNAi therapeutics in cancer therapy and clinical applications
Loutfy H Madkour*
Chemistry Department, Faculty of Science, Tanta University, 31527, Tanta, Egypt
*Address for Correspondence: Loutfy H Madkour, Chemistry Department, Faculty of Science, Tanta University, 31527, Tanta, Egypt, Tel. +201026724286; Email: [email protected]; [email protected]
The RNA interference (RNAi) technique is a new modality for cancer therapy, and several candidates are being tested clinically. Nanotheranostics is a rapidly growing field combining disease diagnosis and therapy, which ultimately may add in the development of ‘personalized medicine’.
Technologies on theranostic nanomedicines has been discussed. We designed and developed bioresponsive and fluorescent hyaluronic acid-iodixanol nanogels (HAI-NGs) for targeted X-ray computed tomography (CT) imaging and chemotherapy of MCF-7 human breast tumors. HAI-NGs were obtained with a small size of ca. 90 nm, bright green fluorescence and high serum stability from hyaluronic acid-cystamine-tetrazole and reductively degradable polyiodixanol-methacrylate via nanoprecipitation and a photo-click crosslinking reaction. This chapter presents an over view of the current status of translating the RNAi cancer therapeutics in the clinic, a brief description of the biological barriers in drug delivery, and the roles of imaging in aspects of administration route, systemic circulation, and cellular barriers for the clinical translation of RNAi cancer therapeutics, and with partial content for discussing the safety concerns. Finally, we focus on imaging-guided delivery of RNAi therapeutics in preclinical development, including the basic principles of different imaging modalities, and their advantages and limitations for biological imaging. With growing number of RNAi therapeutics entering the clinic, various imaging methods will play an important role in facilitating the translation of RNAi cancer therapeutics from bench to bedside.
In the development of RNAi-based therapeutics, imaging methods can provide a visible and quantitative way to investigate the therapeutic effect at anatomical, cellular, andmolecular level; to noninvasively trace the distribution; to and study the biological processes in preclinical and clinical stages. Their abilities are important not only for therapeutic optimization and evaluation but also for shortening of the time of drug development to market. Typically, imaging-functionalized RNAi therapeutics delivery that combines nanovehicles and imaging techniques to study and improve their bio distribution and accumulation in tumor site has been progressively integrated into anticancer drug discovery and development processes.
Since it was discovered that double-stranded RNA (dsRNA) can induce RNA interference (RNAi) in Caenorhabditis elegans [1], the capability of RNAi that can suppress the expression of target genes with high specificity, and efficacy has been confirmed with small RNA sequences (siRNA/miRNA) [2]. Since then, much effort has been directed to develop RNAi therapeutics into clinic as a potential disruptive tool for new conservative and personalized treatment of a broad range of diseases [3], including viral infection [4], eye disorder [5], inflammatory disease [6], cardiovascular disease [7], genetic disorder [8], and cancer [9–11].
Currently, both naked siRNAs and nanoparticle-mediated delivery of RNA fragments have been investigated for cancer therapy. The administration route for naked siRNA is critically reliant on its accessibility to the target sites inside the body. Most of the naked siRNA fragments have been administered topically, which has the advantages of high bioavailability in target organs and low adverse reactions compared to systemic administration. Because the systemic administration of siRNA fragments requires them to reach the right organs or tissues after dispersing in the body and passing through the biological barriers, which is virtually impossible for naked small RNAs. The systemic delivery of siRNAs or other small therapeutic RNAs to target sites is impeded by many factors including nuclease degradation and rapid systemic elimination of “naked” small RNAs in biological systems [12]; intrinsic negative charge nature on the surface of small RNAs make them very difficult to transfer into cells that also with negatively charged surface of bilayer phospholipid membrane [13]. Therefore, a variety of delivery systems have been engineered to address above issues and deliver small RNAs specifically to target sites, including tumors and inflammatory foci, for instance, lipid nanoparticles made with cationic lipids; lipidol and other lipid materials [14–17]; polymeric nanoparticles made with polyethylenimine (PEI); dendrimers; membrane-disruptive and cyclodextrin-containing polymers [18–20]; membrane-disruptive peptides [21,22]; aptamer-siRNA chimeras [23]; single-chain fragmented antibodies [24]; supramolecular assemblies [25–27]; inorganic nanoparticles, including multi shell nanoparticles, mesoporous nanoparticles, and carbon-based nanomaterials [28]; and so on [29–33]. However, the entrapment of nano-sized carriers in the reticuloendothelial system (RES), the vascular endothelial resistance in tissues, the barriers of extracellular matrix, and the intracellular release from endosomal uptake [34,35] are the major challenges for the systemic delivery of RNAi therapeutics. In addition, the siRNA-based therapeutics encounter safety concerns owing to unexpected adverse effects, including off-target effects, innate immune responses, proinflammatory cytokine induction, and spleen toxicity, which were found in the early studies of those candidates [8,36–38].
There are three major aspects of scientific challenges in developing RNAi therapeutics for cancer therapy (Figure 1):
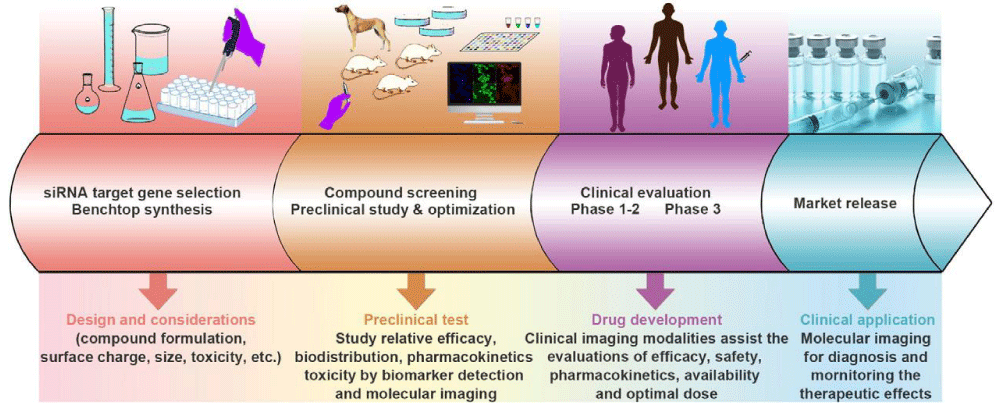
Figure 1: The workflow of developing RNAi therapeutics and translating them for clinical applications. There are mainly four steps for this, including targeting gene selection, compound screening, clinical evaluation, and market release. Imaging methods could assist the development of RNAi therapeutics at every step in the RNAi therapeutics development processes, including marking for gene selection, tracing the small RNA sequences for pharmacokinetics study, evaluating the gene-silencing efficacy, and diagnosing tumors and monitoring the therapeutic effects.
(1) target gene selection for RNAi and gene vector development for either systemic or local administration; (2) product screening and preclinical evaluation including the relative efficacy, bio distribution, pharmacokinetics, and toxicity; and (3) clinical study of the efficacy, safety, pharmacokinetics, and optimal dose. It has been proven that noninvasive imaging methods and biomarker detection could speed up the development of RNAi therapeutics [39–41]. Developing powerful imaging techniques and methods is important for providing valuable information by visualizing, characterizing, and quantifying the biological processes of RNAi therapeutics and monitoring their therapeutic effects and factors that are crucial for the optimization of RNAi cancer therapeutics.
Generally, in vivo imaging of RNAi refers to the utilization of a variety of imaging modalities, including bioluminescence imaging (BLI) [42], photoluminescence imaging [43,44], magnetic resonance imaging (MRI) [45], positron emission tomography (PET) [46], single-photon emission computed tomography (SPECT) [47], and ultrasound [48], to quantitatively and/or qualitatively visualize the in vivo behavior of the RNAi therapeutics. Many imaging modalities have been applied for RNAi researches. The therapeutic genes have been directly or indirectly marked with a variety of imaging contrast agents to make them detectable in biological systems with clinically relevant imaging equipment. These probe-labeled systems with target delivery function hold high promise for studying the pharmacological properties of RNAi therapeutics in clinical translation. This chapter highlights the current status of RNAi-based cancer therapeutics in clinical trials with discussion of selected cases. Before addressing the current progresses of applying various imaging techniques in RNAi therapeutics development, we will discuss the biological barriers and challenges for developing RNAi therapeutics and emphasize the design considerations of non-viral gene delivery vehicles for targeting tumors. Finally, the perspective of applying various imaging techniques for developing RNAi cancer therapeutics is described in more detail.
RNAi cancer therapeutics in clinical trials
The significant advantages of RNAi strategy are its high specificity together with infinite choice of genes that can be applied for cancer therapy. The RNAi therapeutics could interfere with angiogenesis, metastasis, chemo resistance of tumors, and the proliferation of cancer cells [49,50]. The intrinsic features of RNAi therapeutics make RNAi a novel strategy for cancer treatment. Therefore, a myriad of RNAi therapeutics is under investigation for this purpose. Recently, around 10 types of RNAi-based cancer therapeutics have entered the early stage of clinical trial (Table 1), demonstrating potential capability of RNAi with specific gene-silencing efficacy for cancer treatment. Although the ongoing clinical trials provide encouraging results for future commercial success, many obstacles still remain ahead for applying RNAi therapeutics in humans. Here, we highlight the current status of two selected cases in clinical evaluation.
| Table 1: Current clinical status of RNAi therapeutics for cancer treatment. | ||||||
| Indications | Name | Delivery Route | Target | Delivery system | Development phase |
References |
| Advanced solid tumors | siRNA-EphA2-OPC | Intravenous injection | EphA2 | Lipid-based nanoparticles | Preclinical | [51] |
| Metastatic tumors or cannot be removed by surgery | APN401 | Intravenous injection | E3 ubiquitin ligase Cbl-b | Ex vivo transfection | Preclinical | [52] |
| Metastatic melanoma, absence of CNS metastases | iPsiRNA | Intradermal injection | LMP2, LMP7, MECL1 | Ex vivo transfection | Phase I, completed |
[53] |
| Advanced solid tumors | Atu027 | Intravenous infusion | PKN3 | Lipid-based nanoparticles | Phase I, completed |
[54] |
| Pancreatic ductal adenocarcinoma; Pancreatic cancer | siG12D LODER | Intratumoral implantation | KRASG12D | LODER polymer | Phase I, completed |
[55] |
| Primary or secondary liver cancer | TKM-080,301 | Hepatic intra-arterial injection |
PLK1 | Lipid-based nanoparticles | Phase I, completed |
[56] |
| METAVIR F3-4 | ND-L02-s0201 | Intravenous injection | HSP47 | Lipid-based nanoparticles | Phase I, recruiting |
[57] |
| Solid tumors; multiple myeloma; non-Hodgkin's lymphoma |
DCR-MYC | Intravenous infusion | MYC | Lipid-based nanoparticles | Phase I, recruiting |
[58] |
| Cancer; solid tumor | CALAA-01 | Intravenous injection | RRM2 | cyclodextrin-containing polymer |
Phase I, terminated |
[59] |
| Neuroendocrine tumors; adrenocortical carcinoma | TKM 080301 | Intravenous infusion | PLK1 | Lipid-based nanoparticles | Phase I/II, recruiting |
[60] |
| Solid tumors | ALN-VSP02 | Intravenous injection | KSP, VEGF | Lipid-based nanoparticles | Phase I, completed |
[61,62] |
The siRNA-loaded lipid nanoparticles (Atu027) have been applied clinically to suppress the expression of protein kinase N3 (PKN3) [63]. PKN3 is an effector of PI3K pathway related to the modulation of cell growth, differentiation, survival, motility, and adhesion as well as immune cell and glucose transport function. Chronic activation of PI3K pathway could prevent various human cancers and inhibit the growth of malignant cells [64]. Although numerous signaling molecules can be considered as therapeutic candidates to mediate the PI3K pathway, their upstream inhibition could trigger signal cascades with undesirable signal regulation of normal cells associated with various side effects. For this reason, PKN3 is considered to be a proper effector to adjust the growth of metastatic cancer cells with activated PI3K [65]. The results from animal studies indicate that Atu027 could effectively knock down the expression of PKN3 gene in the vascular endothelium to inhibit tumor growth and metastasis [63]. In clinical study of Atu027, no interferon response or activation of cytokines was observed, which may be due to liposomal encapsulation of siRNA with enhanced safety while avoiding triggering side reactions during circulation [66]. Thus, Atu027 was well tolerated, and no dose-dependent toxicity was observed. Recent study of using Atu027 to treat advanced solid tumors has found that up to 41% of patients exhibited no further progression of tumors after eight weeks of treatment [67]. Since Atu027 targets tumor stroma instead of tumor cells, it is expected that this treatment will be effective for all type of vascularized metastatic cancers.
Another clinical trial product that targets kinesin spindle protein (KSP) and vascular endothelial growth factor (VEGF) for treating solid tumors is ALN-VSP02. KSP is a type of motor protein that plays a central role in the proper separation of emerging spindle poles during mitosis, and it is upregulated in many types of cancer cells. Therefore, KSP is another attractive target for cancer therapy by RNAi therapeutics, as silencing its expression will lead to cell cycle arrest at mitosis through the formation of an abnormal mitotic spindle and finally inducing apoptosis [68]. VEGF, which is involved in angiogenesis and lymphangiogenesis, is overexpressed in numerous cancer types [69,70]. Blocking the expression of VEGF is expected to inhibit angiogenesis and suppress tumor growth [71]. The dually targeted RNAi drug (ALN-VSP02) is in clinical trial aimed for treating solid tumors (Figure 2a). In the first-in-human study, the clinical activity, safety, and pharmacokinetics of ALN-VSP02 were evaluated, demonstrating good systemic tolerance and acceptable toxicity with biweekly intravenous administration. The results also demonstrated that the expression levels of both target genes were decreased in multiple patients, including one patient with complete regression of liver metastases from endometrial cancer (Figure 2b,c) [11]. Although ALN-VSP02 has achieved some success in clinical trials, some interesting results should be noticed. The infusion-related reactions (IRR) of ALNVSP02 seem to be complement-mediated, not cytokine-induced. Additionally, it was found that ALN-VSP02 could cause spleen toxicity with prolonged dosing. The patient with endometrial cancer had a more than 50% decrease in blood flow (Ktrans) and a 90% reduction in spleen size [11]. Based on the above observations, some improvements are suggested for future development of Stable nucleic acid lipid particle (SNALP)-based cancer siRNA therapeutics to improve the safety. It includes using more efficacious and less immunogenic lipid components and reducing the size of SNALP-based drug delivery systems to be small enough to improve the circulation time and benefit the accumulation in solid tumors through the enhanced permeability and retention (EPR) effect. Finally, the biodegradability of SNALP should be optimized to reduce the spleen toxicity, which was induced by lipid accumulation along the endosomallysosomal pathway.
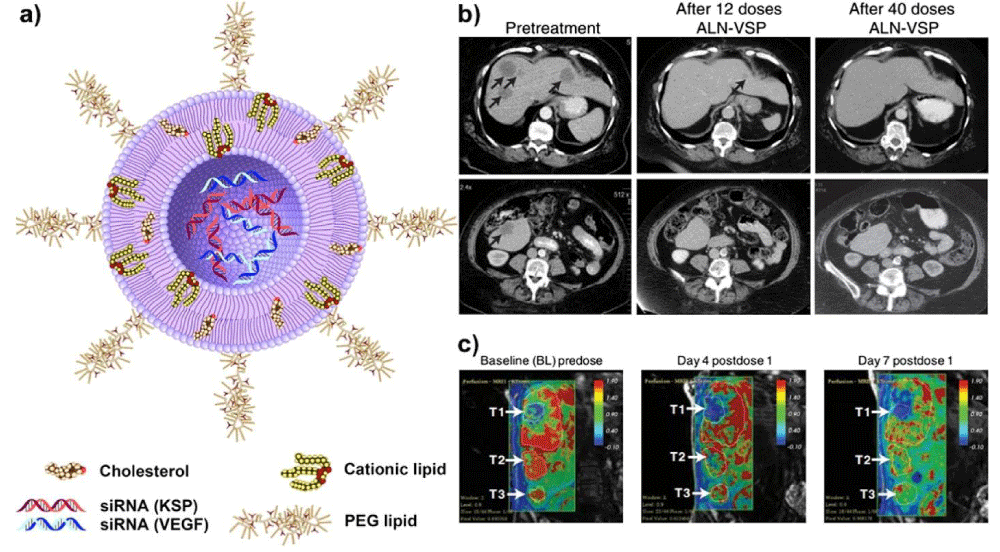
Figure 2: (a) Schematic structure of ALN-VSP formulation: The siRNA lipid nanoparticle is composed of cationic or ionizable lipid with cholesterol and encapsulated with two different siRNAs to target KSP and VEGF genes. The nanoparticles are stabilized by surface binding of PEGylated lipids. (b) CT images demonstrated a complete response of ALN-VSP in an endometrial cancer patient with multiple liver metastatic tumors (black arrows). (c) Dynamic contrast-enhanced MRI (DCE-MRI) images from another patient with three metastatic pancreatic neuroendocrine tumors in the right lobe of the liver (white arrows). A significant change of tumor blood flow was observed during the treatment at 0.7 mg/kg (indicated in red). Figures adapted with permission from [11].
To date, owing to the low stability, poor systemic distribution, possible side reactions, and low bioavailability of naked siRNA, most siRNA based therapeutics in clinical trials are formulated by encapsulation within lipid-based particles. These early trials hold great potential for cancer therapy, especially allowing RNAi to be precisely tailored in each case. However, many challenges still need to be solved for personalized cancer therapy by RNAi to become a reality. First, RNAi delivery systems with high efficiency and low toxicity are needed for early (Phase I) clinical trial. Then an adequate knockdown of target genes needs to be achieved in later trials of Phase I to Phase II. Besides, possible off-target effects in normal tissues should be avoided for safety consideration (Phase II). The positive or negative correlation of target knockdown with tumor regression should be analyzed during Phase II to verify the feasibility for Phase III study. Finally, for future clinical applications, the distribution, metabolism, and degradation of siRNA loaded nanocarriers need to be extensively studied.
Biological barriers for RNAi cancer therapeutics
Although several siRNA-based therapeutics have been evaluated in early phase clinical studies, there are various biological barriers that need to be conquered for successful clinical translation, such as efficient delivery of RNAi therapeutics to tumors after reaching the circulation, overcoming the vascular barrier, cellular uptake, and endosomal escape (Figure 3). Because many target sites are not accessible or not convenient for local administration, thus the systemic administration of RNAi therapeutics is essential. To fully exploit the therapeutic potential of RNAi therapeutics, developing effective and biocompatible gene delivery systems that could specifically target cancer cells in the body is a key factor. Generally, gene vehicles with the diameter ranging from 10 to 100 nm are suitable for systemic administration. These gene vectors are needed to be stable enough and well dispersed in blood. Besides, gene vectors decorated with targeting moieties could facilitate specific cellular uptake by cancer cells, a strategy that is important to evade the innate immune stimulation in the body [72]. In such cases, noninvasive imaging tools can be utilized to study the pharmacokinetic properties of RNAi therapeutics and monitor their effects in a visible way. In order to accelerate the screening process, imaging techniques with adequate spatial resolution and sensitivity for small animals should be used for determining the temporal and spatial bio distribution of the developed compounds. This may help reduce irrational costs and allow the selection of the most promising candidates during the early stage of drug development [73].
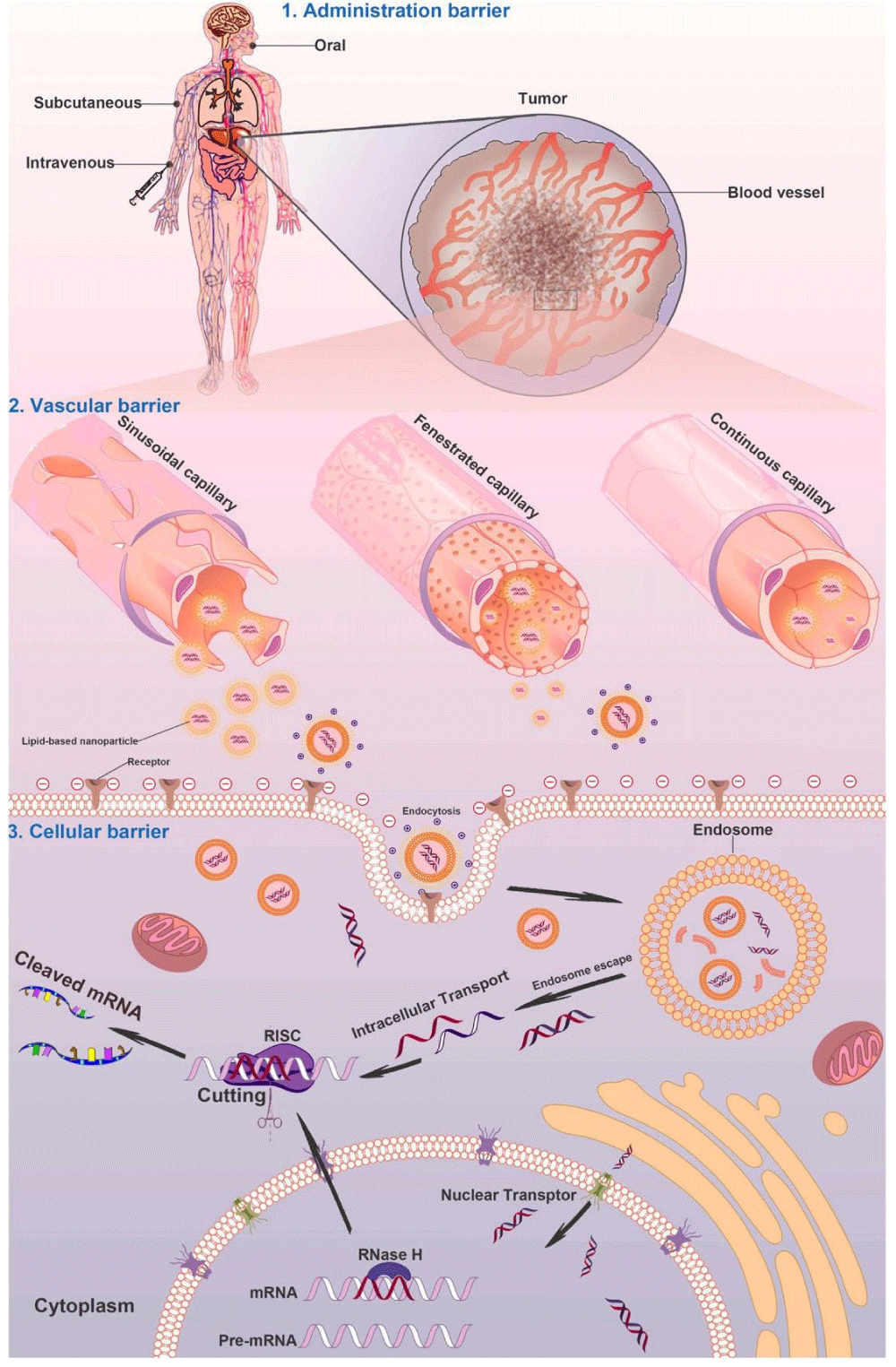
Figure 3: Schematic illustration of nanocarrier-mediated delivery of RNAi therapeutics: the biological barriers for gene silencing in cancer treatment include reaching the circulation, crossing the vascular barrier, cellular uptake, and endosomal escape. Following systemic administration in patients, the RNAi therapeutics could be transported to the blood vessels in tumor tissues. The gene vectors could escape from the sinusoidal and fenestrated capillaries and retain in the tumor regions. The gene vectors could be taken by cancer cells through endocytosis or ligand- mediated intracellular transport and then transferred into the cytoplasm to silence target genes for cancer therapy.
Administration barrier: Administration through oral route is obviously the most convenient approach for patients. However, it is currently challenging for treating cancers through the oral administration of RNAi therapeutics due to difficulties in accessing tumor sites, including poor intestinal stability and insufficient permeability across intestinal epithelium into circulation [34]. Subcutaneous administration is another route for the systemic delivery of RNAi therapeutics. The drugs can reach the circulation directly fromthe interstitial space of subcutaneous connective tissue to the capillaries by traversing through the vascular barrier or through lymphatic drainage. Compared to intravenous administration, the sustained entry of drugs into circulation through subcutaneous route can achieve almost complete absorption without first-pass effect in the liver. A clinical example of RNAi therapeutics using subcutaneous administration to treat transthyretin-mediated amyloidosis is ALN-TTRsc, which could achieve approximately 90% knockdown of the transthyretin gene expression in liver in a phase I study [74]. For the subcutaneous administration of RNAi therapeutics, the lipophilicity and size of gene vectors have to be taken into account to avoid endocytosis by phagocytic cells in subcutis and lymph node drainage, which subsequently influence the potency of RNAi therapeutics. For further details on designing desirable properties of siRNA delivery system, please refer to the following reviews [17,19,75–79]. The most direct way for RNAi therapeutics to reach blood circulation is intravenous or infusion injection. Currently, several RNAi products for systemic administration have entered clinical evaluation for cancer treatment (Table 1). In order to identify and validate gene vector candidates for specific administration route of siRNA delivery during preclinical stage, in vitro and in vivo optical imaging can be assisted as a fast and inexpensive method for compound screening by visualizing and evaluating the biocompatibility, stability, absorption, and distribution in a live subjects in a real-time manner, as well as biological interactions at subcellular level.
Vascular barrier: Passing through the endothelium of vasculature is a key step for RNAi therapeutics. Depending on the vascular permeability of target organs or tissues, a certain half-life of RNAi therapeutics in plasma is required. A successful gene silencing in the liver primarily benefits from the discontinuous sinusoidal capillaries, as the large openings in the endotheliumcan greatly access the leaked RNAi nanocarriers from the vasculature. Such pores in sinusoidal capillaries are wide openings for both passive and active passages of RNAi nanocarriers up to 100 nm in size from bloodstream to hepatocytes in liver. Tumor capillaries are discontinuous, with considerable variation of cell composition, the basement membrane, and pericyte coverage. Kobayashi, et al. suggested that four important factors should be considered when passive targeting is involved [80]: (1) internal and external blood flow of tumor, (2) tumor vascular permeability, (3) structural barriers enforced by extracellular matrix and tumor cells, and (4) intratumoral interstitial pressure. These factors can certainly influence the accumulation of certain sized nanoparticles or molecules in tumor tissue by EPR effect [81]. In order to take advantage of the EPR effect in tumor tissues, or receptor mediated transcytotic pathway through vascular endothelium [34], longer half-life for RNAi therapeutics is necessary. In contrast to sinusoidal or tumor capillaries, the fenestrated capillaries have much smaller pores (60–80 nm in diameter) in endothelium covered with continuous basal lamina, which can prevent the diffusion of large-scale nanoparticles. They are mainly located in the endocrine glands, intestines, pancreas, and glomeruli of kidneys. The tightness, shape of the pores, continuous basal lamina, and extracellular matrix should be considered for designing non-hepatic tissues-targeted RNAi-based formulations and delivery strategies.
The stability of RNAi therapeutics in blood circulation is of primary importance for arriving tumor tissues. A major challenge is to evade the phagocytic uptake by mononuclear phagocyte system (MPS) in the bloodstream [82]. The formulation size, surface electrostatic nature, and stability can certainly affect the uptake by MPS. It is more likely to interact with MPS and other components in the bloodstream if they possess large size and excessive net charge [83]. Thus, the formulations are generally of minimum net charge through modification with hydrophilic and neutral molecules to increase the stability. Introducing poly(ethylene glycol) (PEG) shell to the surface of RNAi vectors is a general approach for stabilization. However, the high stability achieved by PEGylation can reduce uptake by target cells [84]. This can be solved by attaching targeting moieties to RNAi therapeutics [85]. The vascular endothelium is negatively charged because the heparin sulfate proteoglycans are on the cell surface and in the extra cellular matrix. Additionally, tumor vascularity is controlled by oxygen supply and some metabolites [86]. It has been observed that increasing the tumor vascularity could result in more efficient delivery of RNAi agents to solid tumors [87]. It was reported that nanoparticles with smaller size and increased lipophilicity could lead to higher level of accumulation in tumor tissues [88]. To evaluate the delivery efficiency and in vivo behavior of the potential RNAi therapeutics, quantitative functional, and molecular imaging can be integrated to provide valuable information concerning pharmacokinetics and bio distribution properties in animal models, such as quantitative visualization of compound absorption and distribution in tissues and organs, as well as elimination time after the administration, and the amount of compounds that reaching target tissues. Usually, the quantitative imaging assessment can be performed in a real-time, whole-body, and noninvasive way by selecting appropriate imaging techniques. In this method, the compound is often labeled with specific imaging probes corresponding to the imaging modality for further applications. A great benefit of this approach is that the animals can be imaged repeatedly for longitudinal studies, which minimizes the number of animals needed for a given experiment [89].
Cellular barrier: Cellular uptake is critical for successful delivery of RNAi therapeutics into the cytoplasm for gene silencing. Basically, cell membrane is comprised of hydrophobic phospholipid bilayer embedded with various functional proteins. The innate negatively charged cell surface provides an external biological barrier to naked siRNA molecules. The small cationic peptides, cationic lipids, and polymers have often been applied to facilitate the cellular uptake of siRNA molecules via endocytosis [90]. However, for targeted delivery of RNAi therapeutics into cancer cells, the receptor-mediated endocytosis is mostly preferred, and various ligands have been used for targeted delivery of RNAi therapeutics, including folate [91], transferrin [92], and aptamers [93]. Those ligands could specifically interact with receptors overexpressed on the surface of cancer cells to promote cellular uptake. Pros and cons of using various targeting ligands for siRNA therapeutics have been reviewed elsewhere [94]. Once entering into target cells, the success in approaching RNA induced silencing complex (RISC) in cytoplasm is largely dependent on endosomal escape. It has been suggested that endosomal escape should occur before late endosomes fuse with lysosomes, which contains certain digestive enzymes [95]. For cationic polymers, which could enhance endosomolysis via absorbing protons and preventing the acidification of the endosomes, the elevated influx of the protons to endosomes increases osmotic pressure that causes lysosome swelling and rupture, eventually releasing the gene vectors to cytoplasm [96,97]. Similarly, ionizable lipids with neutral charge in bloodstream can become positively charged in endosomes, subsequently leading to disruption of the endosome membrane [17]. Elucidating the mechanism of endosomal escape pathways can help develop new strategies for gene delivery without relying on acidification. For investigations of cellular uptake and intracellular trafficking, molecular/cellular imaging is an essential tool to demonstrate the ligand-receptor interaction, subcellular translocation, and mechanism of potential RNAi therapeutics. Fluorescent probes and radioactive isotope-labeled drug candidates are commonly used for whole-body and subcellular tracking to provide quantitative and qualitative information of biological processes occurring at cellular and molecular levels.
Immune response and safety: The key issue in the application of RNAi therapeutic is the safety without any undesirable side effects. Unwanted silencing of target genes in normal organs or tissues is called “off-target silencing” [38,98]. The siRNAs longer than 30 bp can induce the interferon pathway [36]. Such induction of interferon reaction is caused by the innate immune system because the human body recognizes long dsRNA as virus particles and triggers the innate immune system to overcome the infection. Several investigations showed that even low concentration of siRNAs can induce natural immunity by activating interferon expression [99]. It is suggested that the main mechanisms of immune response caused by some siRNAs are the stimulated production of proinflammatory cytokines through TLR-8 on monocytes and TLR-7 on dendritic cells in a sequence-dependent manner [36,100]. In order to overcome this problem, the therapeutic siRNAs should be less than 21–23 bp [36]. Chemicalmodifications including 2′-O-methylation are also performed to avoid immune activation of siRNAs. Hence, immunostimulatory effects of potential siRNA-based therapeutics must be evaluated in animals prior to the clinical trial until the exact mechanism of sequence-dependent siRNA-induced immune response is fully understood [101].
In addition to siRNA-induced immune activation, the formulation of siRNA-based nanoparticles also plays an important role in safety profile for systemic delivery. Presently, the collective results of siRNA SNALP-based nanoparticles (ALN-VSP), cationic liposome/lipoplexes (Atu027), and cyclodextrin-based polymers (CALAA-01) have shown RNAi efficacy and dose tolerability in early clinical development. Based on these valuable outcomes and lessons learned, several additional key investigations are suggested here. (1) The quality control assay for RNAi products must be performed to prevent structure alteration during the practical use. (2) A better understanding of proinflammatory cytokine response is needed for developing more potent siRNA-based nanoparticles with less acute immunostimulatory events. The side effects could be reflected in the change of pathological states or pathological indicators, such as inflammation, enzyme levels, and other biological factors. (3) Imaging techniques could be applied to study the safety of RNAi therapeutics. Once the safety evaluation of a potential RNAi therapeutic has been established in early trial, molecular imaging can assist in the establishment of biological activity at appropriate dosage range with acceptable toxicities associated with the detection, diagnosis, evaluation, treatment, and management of cancer.
Imaging modalities in the RNAi cancer therapeutics development process
Noninvasive imaging techniques are important tools to visualize and quantify the biological processes of RNAi therapeutics at cellular or tissue levels in a real-time way. Imaging-guided delivery of RNAi therapeutics can trace the pathway of RNAi therapeutics inside the body, provide pathological information of tumors, evaluate the tumor targeted delivery, and provide further information about the pharmacokinetics of the RNAi therapeutics.
Optical imaging: Optical imaging, mainly including fluorescence imaging and bioluminescence imaging (BLI), which provides the most convenient way for preclinical study of RNAi knockdown due to their advantages including abundant choices of optical dyes, easy labeling, noninvasiveness, multi-channel imaging function, and whole-body real-time readout. Besides, the excellent sensitivity and inexpensive use of optical imaging (with maximum penetration depth of few centimeters [102,103]) make it a promising modality for real-time monitoring of siRNA delivery in small animals.
For fluorescence imaging, the animals are illuminated by a light at an appropriate wavelength to excite the fluorescent agents in situ, and then the emitted light from fluorescent agents is filtered and detected by a charge-coupled device (CCD) camera maintained at low temperature (Figure 4a) [104]. Since both the excitation and emission lights used are low-energy photons, it is considerably safer than other imaging systems that involve ionizing radiation. However, the use of relatively low energy light results in limited tissue penetration due to the light absorption by tissue components, which makes it virtually impossible for deep tissue imaging in large animals or human subjects. Moreover, there are other intrinsic limitations for consideration, for example, the light scattering phenomenon during the light propagation in tissues can result in a blurry fluorescence image [105]. For in vivo optical imaging, auto fluorescence is an undesired background signals emitted by natural fluorophores in tissues, which can overlay with fluorescent signal from optical probes, and consequently reduce the signal-to-noise ratio [105]. Near-infrared fluorescent (NIRF) agents are generally applied for labeling siRNA therapeutics, as NIRF emits light in the range of 650–900 nm with deep tissue penetration by minimizing the tissue absorption, scattering, and auto fluorescence [106]. For instance, VEGF siRNAs labeled with cyanines dye (Cy5.5) were conjugated to PEG through disulfides to form micelles through the interaction with PEI [107]. With Cy5.5, the VEGF siRNA-loaded micelles could be traced for delivering siRNA into prostate cancer cells (PC3), and the accumulation of Cy5.5-labeled siRNA, siRNA/PEI mixture and micelles could be detected in tumors and the main organs also could be monitored after administration to PC-3 tumor-bearing mice. The fluorescent dyes could also be utilized to study the intracellular pathway of RNAi therapeutics. By image-based analysis of lipid nanoparticles (LNPs) incorporating A647-labeled siRNA, it was observed that the LNPs could enter cancer cells through clathrin-mediated endocytosis and macropinocytosis, while the efficiency of siRNA escape from endosome to cytosol was low (1–2%) and the endosome escape only occurred when the LNPs were located in the compartment sharing early and late endosomal characteristics [108]. In addition, fluorescence imaging could provide multi-channel imaging functions by labeling RNAi therapeutics with multiple dyes. For instance, the Cy3 and Cy5-labeled siRNAs were covalently linked to PEG to formnanocarrier-like loop, which could generate signals for dual imaging of the products inside cancer cells based on the fluorescence resonance energy transfer (FRET) of the fluorophore pair [109]. Furthermore, the RNAi therapeutics in blood circulation, leakage from blood to tumor tissues as well as distribution in tumor tissues can be monitored in a real-time manner with intravital confocal laser scanning microscopy (IVRTCLSM). For instance, the Cy5-siRNAincorporated polymeric micelles and naked siRNA (Cy5-siRNA) could be traced in blood circulation to study their pharmacokinetics in vivo, to investigate their entry from blood to tumor tissues, as well as their distribution in tumor tissues by utilizing IVRTCLSM (Figure 4b,c,d) [110–112].
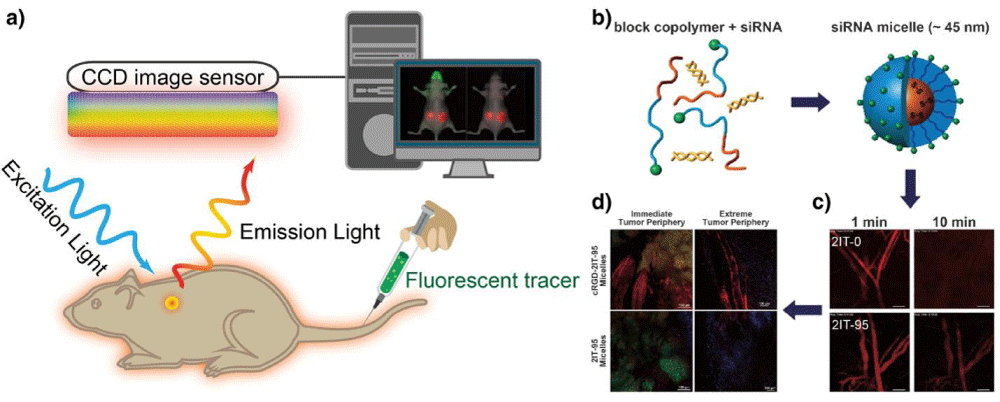
Figure 4: Optical fluorescence imaging. (a) A schematic illustration of the basic principle of in vivo fluorescence imaging. First, a fluorescently labeled RNAi therapeutics is injected into mouse body, and the subject is illuminated by an excitation light at specific wavelength, resulting an excitation of fluorescent molecules and subsequent emission light of different wavelength. The emitted light is filtered and then detected by CCD image sensor. The fluorescence signals and animal photo are finally converted to a single detailed image. The figures on right show using fluorescence imaging to trace polymeric micelles incorporated with dye-labeled siRNA in the mice body. (b) Schematic illustration of polymeric micelle structure formed by self-assembly of block copolymers with fluorescent dye-labeled siRNA. (c) Ear-lobe dermis snap-shots of blood circulation of two types of micelles after intravenous injection for 1 and 10 min. (d) Micro distribution of micelles incorporated with Cy5-labeled siRNA at 24 h post-injection. (red: siRNA; green: tumor cell nuclei; purple: other cell nuclei). Figures adapted with permission from [110].
Besides organic fluorescent dyes, other nanomaterials, such as quantum dots (QDs) [113], carbon dots (C-dots) [114], and up-converting nanoparticles (UCNPs) [115], are also used for optical imaging as well as for siRNA delivery due to their high signal intensity and photostability. Specifically, tumor-specific, multifunctional siRNA loaded QDs were prepared to induce downregulation of the expression of epidermal growth factor receptor variant III (EGFRvIII), which plays an important role in interfering with the proliferation of various types of cancer cells, while the uptake of siRNA-QDs was monitored by fluorescence imaging [116]. Compared to QDs, the C-dots show advantages of better biocompatibility, lower cost, and easier preparation and are considered as a potential alternative to QDs [117]. The C-dots surface coated with alkyl–PEI2k could effectively deliver siRNA and transfect against firefly luciferase (fLuc) with inhibited expression of luciferase gene in 4 T1-luc cells, while maintaining their biocompatibility and fluorescence properties [118]. The UCNPs have gained increasing attention in recent years as a new generation of biological luminescent nanoprobes for cell labeling and optical imaging. It offers many advantages, such as deep penetration, low background auto fluorescence, and high resistance to photo-bleaching, thus providing a promising optical imaging way for monitoring the siRNA transfection [119,120].
In contrast to fluorescence imaging, BLI does not require an external light illumination on the living subjects because the bioluminescence is produced by oxidation reaction of luciferin with catalytic assistance of luciferase enzyme in the body. Various luciferase genes can be introduced into biological systems via transfection techniques and expressed with corresponding luciferase enzymes (Figure 5a). In this technique, there is no interference from auto fluorescence and endogenous bioluminescence. Thus, BLI can provide a higher signal sensitivity and better signal-to-noise ratio than fluorescence imaging. By exposing RNAi therapeutics to cancer cells or injecting to tumor-bearing animals, the expression of bioluminescent reporters and fluorescent proteins, such as luciferases, the green fluorescent protein (GFP), and red fluorescent protein (RFP) [121], could be tested to evaluate the gene-silencing efficacy of those products. This could be helpful in forecasting their interfering ability for screening the most effective siRNA therapeutics and the best formulations before further test in human. In one study, Kay’s group demonstrated the feasibility of monitoring siRNA delivery and assessing silencing effect by in vivo BLI (Figure 5b,c) [122]. In another study, by co-injection of luciferase plasmid and synthetic luciferase siRNA, the silencing effect in a variety of organs was monitored through BLI [123].Moreover, BLI was applied to assess the silencing of the activity of P-glycoprotein (Pgp), a multidrug resistance (MDR1) gene product overexpressed in multidrug-resistant cancer cells by using short hairpin RNA interference (shRNAi) [124]. The shRNAi-mediated downregulation of Pgp activity at cellular level or in animal models could be directly traced by BLI of Renilla luciferase (rLuc) reporter through its substrate, coelenterazine, which is also a known as substrate for Pgp transportation. Furthermore, the in vivo gene-silencing activity of luciferase siRNAs incorporated in calcium phosphate (CaP) nanoparticles was tested in a fLuc-expressing human cervical cancer cell line (HeLa-Luc). These nanoparticles were tested in transgenic mice (FVB/NJc1 female mice) with spontaneous pancreatic tumors by measuring the bioluminescence intensity with IVIS® after intraperitoneal injection of luciferin [125]. Recently, the kinetics of siRNA-mediated gene silencing combined with BLI was studied for the assessment of the best approach for gene silencing [126], for simulating/predicting the effective siRNA dose based on luciferase knockdown in vitro, and for studying the kinetics of luciferase knockdown by RNAi therapeutics in subcutaneous tumors and their effects.
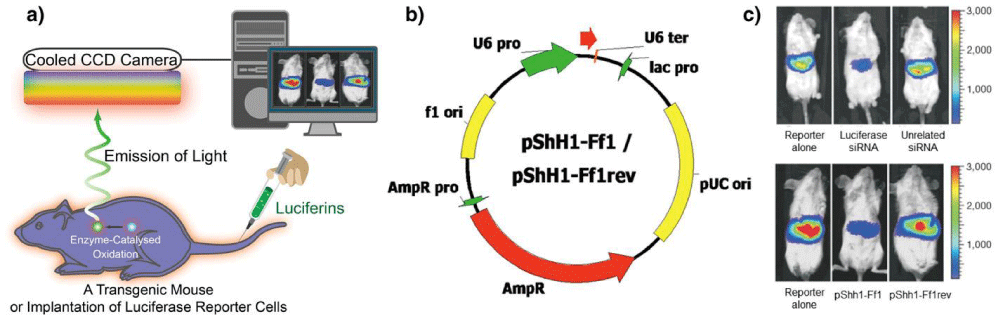
Figure 5: Optical bioluminescence imaging. (a) A schematic illustration of the basic principle of in vivo bioluminescence imaging. In this case, mouse with luciferase (Luc)-labeled cancer cells that express luciferase is required. After injecting the enzyme substrate luciferin into the mouse body, the bioluminescence light is generated when the luciferin molecules interact with luciferase via enzyme-catalyzed oxidation. Finally, the emitted light is detected by a cooled CCD camera and the image is produced by computer. (b) The plasmids used for transfection into mouse liver. (c) Representative images (upper image) of mice co-transfected with luciferase plasmid pGL3-control without siRNA, with luciferase siRNA or with unrelated siRNA. The results indicate that the luciferase expression was specifically suppressed by siRNA-mediated inhibition in adult mice, but the unrelated siRNAs had no effect. Another result from (c) (lower image) shows the gene silencing of luciferase expression by functional shRNAs (pShh1-Ff1). These outcomes demonstrated that the plasmid-encoded shRNAs can induce an effective and specific RNAi response in vivo. Adapted with permission from [122].
GFP and its derivatives have also been widely utilized for imaging in vitro gene silencing of siRNA in numerous studies [127,128]. For instance, silica-gold nanoshells were covalently decorated with epilayer of poly(L-lysine) peptide (PLL) on the surface to load single-stranded antisense DNA oligonucleotides or double-stranded short-interfering RNA (siRNA) molecules with NIR laser irradiation-triggered release of gene segments and endosomal escape [128]. The gene-silencing efficacy was evaluated by measuring the downregulation of GFP in human lung cancer H1299 GFP/RFP cell line. Cancer cells and tumor-bearing animals were used to study the gene-silencing effects for screening the RNAi products and serve as a real-time tool to investigate the efficacy of siRNA delivery in preclinical studies. Understandably, the application of these technologies in humans is limited because of need of reporter gene transfection.
PET and SPECT: PET and SPECT are nuclear imaging techniques that use radioactive tracers and detect gamma rays to provide information of molecular signatures at molecular level within living subjects (Figure 6). Both imaging systems could afford excellent penetration depth in tissues and high sensitivity for whole-body imaging. For PET imaging, the specifically radiolabeled imaging agents are required for targeting and visualizing organs or tissues of interest. Imaging of radioactive tracer is achieved by detecting the high-energy photons (gamma ray) emitted from the radioactive isotopes during the spontaneous radioactive decay. More specifically, the nucleus of specific radioactive isotope undergoes a beta plus (β+) decay due to its unstable nuclear system, while an excessive proton is converted into a neutron, a positron, and an electron neutrino [129]. Based on electron-positron annihilation, the collision between electron and positron produces two gamma ray photons, traveling at opposite directions at approximately 180° from each other [130,131]. The gamma ray has ten times higher energy than X-ray, and large number of emitted paired photons from radioactive isotopes detected by gamma cameras provide angular and radial distance information from regional interest [131]. These features enable high signal sensitivity and reconstruction of quantitative tomographic images.
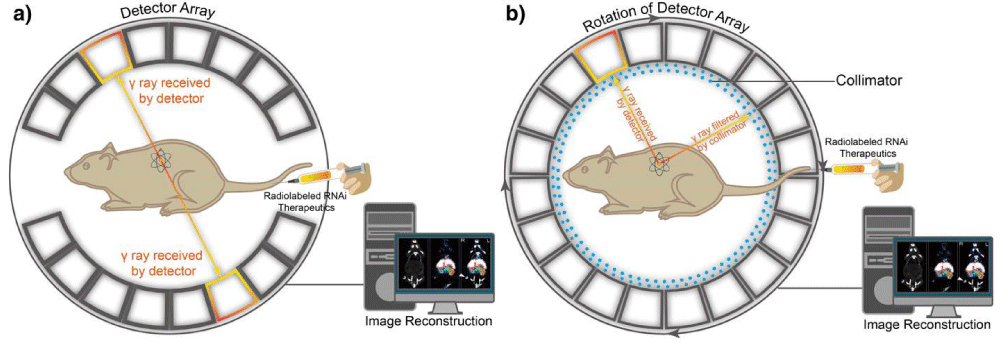
Figure 6: Positron emission tomography (PET) and single-photon emission computed tomography (SPECT). (a) A schematic illustration of the basic principles of in vivo PET imaging. In this technique, the radioactive isotope-labeled RNAi therapeutics is injected into animals. Positrons are emitted from the isotopes associating with electrons, which cause annihilation and subsequent production of two gamma (γ) rays. The two high-energy γ rays are traveling at 180° from each other. Then the γ rays are received by detector array with electrical signals and finally converted into tomographic images. (b) Another schematic illustration of the basic principle of in vivo SPECT imaging. First, the radioactive isotopes-labeled RNAi therapeutics is administered into the mouse to emit γ rays. The γ rays produced by isotopes in SPECT do not travel in opposite directions, instead, are collected by detector array that rotates around animals, while any diagonally incident γ rays are filtered by collimator. The γ rays received by detector array are converted and reconstructed into tomographic images.
Owing to their highly quantitative and sensitive nature, radionuclide imaging techniques have been utilized for analyzing the pharmacokinetics and bio distribution of RNAi therapeutics [132]. Relatively short half-lived isotopes such as 18F (T1/2 = 109.8 min) and 64Cu (T1/2 =12.7 h) are frequently used as radioactive tracers to label siRNA molecules or drug delivery systems for PET imaging. In one recent study, core/shell-structured hollow gold nanospheres (HAuNS) were developed as a targeted NIR light-inducible delivery system for nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) targeting siRNA [133]. By conjugating 1,4,7,10-tetraazacyclododecane-1,4,7,10- tetraacetic acid (DOTA) derivatives to the surface of nanospheres and labeling with 64Cu, the HAuNS were applied for micro-PET/computed tomography (CT) imaging. The PET/CT images indicated that targeted HAuNS showed higher accumulation in tumors than nontargeted nanocarriers in HeLa cervical cancer bearing nude mice after intravenous injection (Figure 7). 18F-labeled siRNA has also been investigated using PET to measure the pharmacokinetics and bio distribution of siRNA delivery systems [134]. For example, Oku et al. used Nsuccinimidyl 4-18F-fluorobenzoate (18F-SFB) to label siRNA for real-time analysis of siRNA delivery [135]. PET images revealed that naked 18F-labeled siRNA was cleared quite rapidly from the blood stream and excreted from the kidneys. However, the cationic liposome/18F-labeled siRNA complexes tended to accumulate in the lung. There is an urgent need to develop facile and efficient 18F-labeling methods for PET imaging of RNAi because most traditional 18F-labeling strategies are time-consuming with low yield.
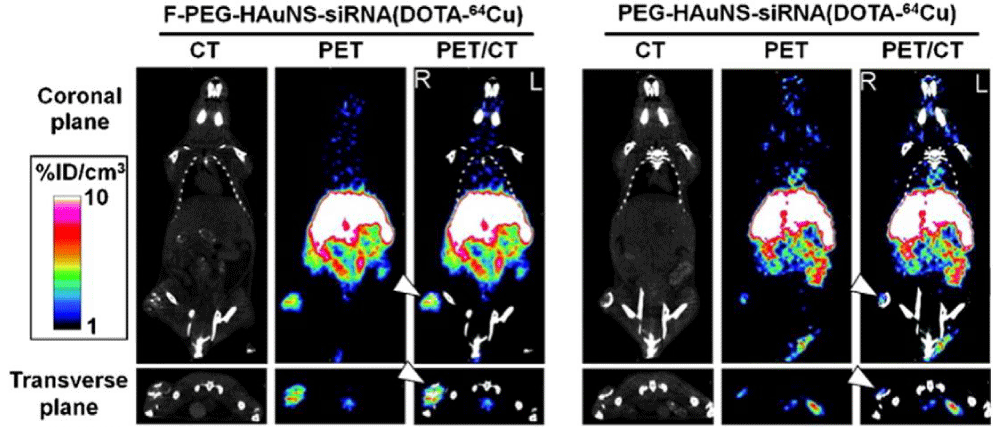
Figure 7: Micro-PET/CT images of nudemice bearingwith HeLa cervical tumor xenografts in the right rear leg at 6 h after intravenous injection of folic acid (F-PEG-HAuNS-siRNA (DOTA-64Cu) or PEG-HAuNS-siRNA (DOTA-64Cu). Tumors were marked by arrowheads. Figure adapted with permission from [133].
Single-photon emission computed tomography (SPECT) is similar to PET by utilizing radioactive materials that decay through the emission of single gamma rays (Figure 6). By comparison, SPECT scans are significantly less expensive than PET since the cyclotron is not required to generate short half-life radioisotopes [136]. SPECT uses isotopes with longer half-lives or from generator elution, such as 111In (T1/2 = 2.8 days), 99mTc (T1/2 = 6h), 123I (T1/2 = 13.3 h), and 131I (T1/2 = 8 days), to provide information about localized function in internal organs with view of the distribution of radionuclides. However, as the emission of gamma rays cannot provide sufficient spatial information for tomographic reconstruction, a special instrumental design for data acquisition is required, and the sensitivity of SPECT can be over 1 order of magnitude lower than PET [136]. In a recent study, siRNA was modified with hydrazinonicotinamide (HYNIC), a chelator for technetium-99m (99mTc), to monitor siRNA at cellular level by gamma counting and micro autoradiography [137]. Besides, the delivery process and bio distribution in tumor-bearing mice were assessed by whole-body imaging. Merkel, et al. also employed SPECT to monitor the bio distribution and pharmacokinetics of siRNA labeled with a gamma emitter (e.g., 111In/99mTc) [138]. In the real-time perfusion investigation, rapid accumulation of gamma emitter-labeled siRNA in the liver and kidneys could be observed, followed by an increasing signal in the bladder. Quantification of scintillation counts in the regions of interest (ROI) revealed that the half-life of siRNA complexes in the blood pool is less than 3 min, suggesting a very rapid excretion into the bladder. Once the siRNA is labeled with radioactive probes, it can be used in noninvasive perfusion, kinetics, and bio distribution evaluation. This offers the advantage of real-time live imaging and investigation at various time points in the same animal to reduce the number of animals needed compared to conventional methods.
MRI: MRI is an important versatile technique that provides noninvasive imaging based on the principle of nuclear magnetic resonance (NMR), by using strongmagnetic field and radiofrequency (RF) pulses to generate RF signal (relying on intrinsic physiological feature) for visualization (Figure 8a). Specifically, an atom nucleus consists of a number of protons and neutrons, each of which has a constant spin and produces angular momentum, which consequently leads to a net angular momentum in the nucleus. If there is an equal number of protons and neutrons in nucleus, net angular momentum is zero. If there is an unequal number of protons and neutrons, then the nucleus gives a specific net spin angular momentum. In the latter circumstances, the nuclear Larmor precession is gained when an external magnetic field is present, and the resonant absorption of RF pulses by nucleus will occur when the frequency of RF pulses equals to the Larmor precession rate. Finally, the RF signal is generated after the removal of external magnetic field [139]. In this regard, the 1H nucleus is particularly useful for MRI since it is abundant in aqueous physiological environment and is magnetically active to give a large magnetic moment to generate RF. However, the RF signal can only be detected from an excess of nuclei with spins aligned either parallel or anti-parallel direction, an equal number of nuclei spins pointing in opposite direction cannot generate detectable MR signals [129]. Therefore, MRI is limited by low sensitivity with long signal acquisition time. Nonetheless, MRI has a number of unique advantages including high spatial resolution, deep tissue penetration, and excellent soft tissue contrast. MRI has been widely used in the clinic to study the anatomy as well as function of tissues. In addition to the development of high field scanners, the design of contrast agents (CAs) plays an important role to improve the image quality by enhancing the contrast of diseased regions while sparing normal tissues. Generally, the CAs could be classified as T1 and T2 CAs due to their magnetic properties and relaxation mechanisms (Figure 8b).
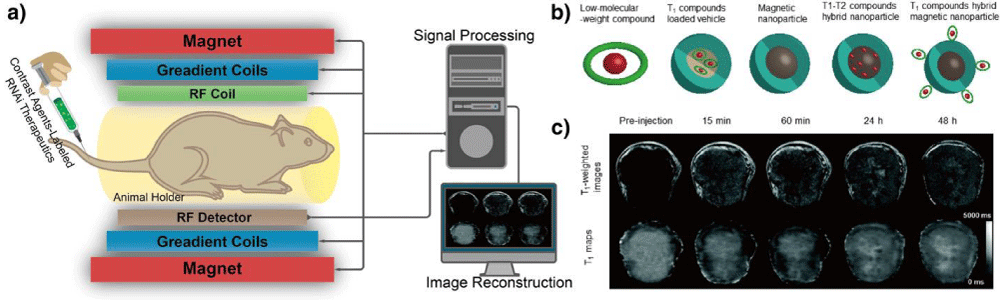
Figure 8: Magnetic resonance imaging (MRI). (a) A schematic illustration of the basic principle of in vivo MR imaging. In general, an MRI scanner consists of three types of coils: the first coil provides a strong homogenous magnetic field, the second coil generates the varying strength of magnetic field in X, Y, and Z directions to encode the spatial position of MR signal and the third coil produces the radio frequency to alter the magnetic dipoles of protons in the subject, generating MR signals to be detected and reconstructed into MR image by computer. MR contrast agents can be labeled onto RNAi therapeutics to track their distribution in vivo through T1 and T2 signal enhancement. (b) Typical contrast agents for MRI, including low molecular-weight paramagnetic compound, T1 CA-loaded vehicle, magnetic nanoparticle, T1-T2 compounds hybrid nanoparticle, and T1 compounds hybrid magnetic nanoparticle. (c) Representative T1-weighted MR images (top) and quantitative T1 maps (down) of a tumor (400 mm3) before and after intravenous injection of siRNA-incorporated nanoplex at the dose of 300 mg/kg. Figures adapted with permission from [143].
Super paramagnetic iron oxide nanoparticles (SPIONs) have the ability to decrease the spin–spin relaxation time for T2-weighted imaging of specific tissues. There are several types of SPIONs approved as contrast agent’s for MRI in the clinic [140]. Recently, Mok, et al. designed and synthesized a pH-sensitive siRNA-loaded nanovector based on SPIONs. The SPIONs were modified with PEI, a commonly used gene transfection macromolecule, through acid-cleavable citraconic anhydride bonds, and coated with anti-GFP siRNA and tumor-specific ligand, chlorotoxin (CTX) [141]. The nanovectors exhibited excellent magnetic property for MRI with a significantly higher r2 (673 mM−1 s−1) than the commercial available T2 contrast agents (e.g., Feridex). More interestingly, the nanovectors did not elicit obvious cytotoxicity at pH 7.4, but exhibited significant cytotoxicity at pH 6.2 as a result of acidic environment elicited cytotoxicity, which may be caused by the protonation of the primary amine at low pH. Meanwhile, the gene-silencing effect under acidic pH condition was significantly higher than that under physiological pH condition, because the surface of nanoparticles was nearly 3 times more negatively charged at pH 7.4 than that at pH 6.2. In another study, the formulation of polyethylene glycol-graft-polyethylenimine (PEG-g-PEI)-coated SPIONs were prepared, which was further modified with neuroblastoma cell specific disialoganglioside GD2 single-chain antibody fragment [142]. The nanocarriers could deliver Bcl-2 siRNA to cancer cells and knock down the expression of Bcl-2 mRNA. In addition, effective delivery of siRNA was confirmed through the in vitro and in vivo MR imaging studies.
Besides T2 CAs, paramagnetic compounds, such as gadolinium and manganese-based compounds, can elevate the relaxation potential by reducing the T1 relaxation time, which are widely applied as T1 contrast agents. Recently, a type of nanoplex that self-assembled from fluorescein isothiocyanate (FITC)-labeled siRNA-chk duplexes and rhodaminelabeled PEI, in which the PEI segments was linked to poly-L-lysine (PLL) with dual-labeling of Cy5.5 and Gd-DOTA, while the PLL-end was combined with prodrug enzyme bacterial cytosine deaminase (bCD) that can convert the nontoxic prodrug 5-fluorocytosine (5-FC) to cytotoxic 5-fluorouracil (5-FU), for imaging-guided RNAi cancer therapy [143]. The nanoplex labeled with different types of CAs could make it possible for MR and optical imaging of the delivery of siRNA and the function of prodrug enzyme in breast tumors for image-guided andmolecular targeted cancer therapy. For instance, the high-resolution T1-weighted MR images and quantitative T1 map of tumor region were obtained by MRI, and the contrast in tumor region was enhanced after the administration of nanoplex (Figure 8c) with T1 value change as a result of the accumulation and diffusion of the nanoplex, demonstrating successful delivery of siRNA into tumor tissues also confirmed by optical imaging. The delivered siRNA could downregulate the activity of aggressive enzyme of choline kinase-α (Chk-α) in breast cancer cells, while the bCD could convert 5-FC to 5-FU, which procedure could be noninvasively monitored by 1HMR spectroscopic imaging and 19F MR spectroscopy. In another study, hollow manganese oxide nanoparticles (HMONs) were exploited as a theranostic Nano platform for simultaneous cancer targeted siRNA delivery and MR imaging [144]. In this study, HMON nanoparticles were coupled with 3,4-dihydroxy-Lphenylalanine conjugated branched PEI (PEI-DOPA) through the strong affinity between DOPA and metal oxides and further modified with Herceptin, a therapeutic monoclonal antibody to target Her-2 expressing cancer cells selectively. Although SPIONs have already been widely used as T2 MRI contrast agents, they still have some drawbacks, such as magnetic susceptibility artifacts and negative contrast, which limit their clinical applications [145]. The development of T1-T2 dual-modal contrast agents has attracted considerable interest because they can provide the contrast for T1-weighted imaging with high tissue resolution and for T2-weighted imaging with high feasibility of lesions detection. Recently, Wang, et al. reported a low-molecular-weight polyethylenimine (stPEI)-wrapped and gadolinium-embedded iron oxide (GdIO) nanoclusters (GdIO–stPEI) for T1–T2 dual-modal MRIvisible siRNA delivery, which exhibited high relaxivities for MRI measurements and suppressed expression of luciferase proteins for dualtype of MR imaging-guided siRNA delivery [146].
Ultrasound: Ultrasound is a clinically widely equipped imaging modality for evaluating the structure, function, and blood flow of organs, which could provide images with high spatial and temporal resolution at low cost (Figure 9a). Ultrasound scanners can emit sound waves with frequencies between 1 and 20 MHz and receive feedback waves reflected by tissues based on density difference to build images for diagnosis [147], which could provide images in a real-time manner without processing delay after acquisition compared with other imaging modalities [40]. In principle, the signal reflected from tissues is insufficient for precise diagnosis because of artifacts from normal tissues. Thus, ultrasound contrast agents are essential to increase the imaging accuracy. Some contrast agents have been developed to enhance positive signal for ultrasound imaging [148–150], such as microbubbles [151–153], nanodroplets [154–157], nanobubbles [158,159], and liposomes [160,161] (Figure 9b).
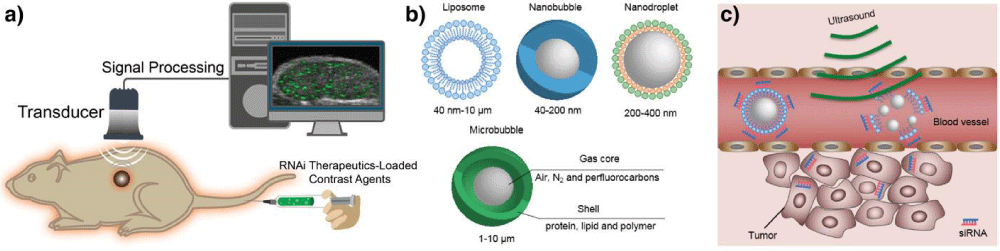
Figure 9: Ultrasound imaging. (a) A schematic illustration of the basic principle of in vivo ultrasound imaging. First, the ultrasound contrast agent that encapsulates RNAi therapeutics is injected into animals, then high-frequency sound waves are sent and penetrated into the subjects, while the time intervals of subsequent reflection of sound waves is recorded by a transducer. The detected signals are converted and constructed into images. A coupling medium is usually used between the contact surface of transducer and subject for sound wave transmission. (b) Typical contrast agents for ultrasound, such as liposome, nanobubble, nanodroplet, and microbubble. (c) Ultrasound-triggered destruction of siRNA-incorporated microbubbles. Ultrasound could disrupt the contrast agents with high acoustic pressure to release RNAi agents when delivered to the blood vessels of the tumor region. Then the released RNAi agents enter into tumor cells to silence target gene.
These are usually constructed with shell of proteins, polymers, lipids, or surfactants to maintain stability in the bloodstream as well as escaping from RES,while loading air or biologically inert heavy gas such as nitrogen, perfluorocarbons, and sulfur hexafluoride to generate echogenicity (Figure 9b) [162]. Besides, solid nanoparticles with cavities that can trap gas [163], and nanoparticles constructedwith gas generating materials have also been applied to enhance the contrast for ultrasound imaging [164].
Ultrasound demonstrates potential advantages in the development of RNAi therapeutics. First, with low acoustic pressure (< 100 kPa) when the ultrasound probe arrives the tumor vasculature, ultrasound could be used to diagnose tumors for imaging-guided delivery of RNAi therapeutics and monitoring the therapeutic effects. Besides, by applying high acoustic pressure (100 kPa to several MPa), ultrasound could be applied to disrupt the probes to release cargos (drugs or RNAs) in target positions and change the permeability of cell membrane with more siRNA delivered intracellularly for gene silencing [152]. As a result, ultrasound can enhance therapeutic effect of RNAi therapeutics (Figure 9c) [149,165]. The siRNA molecules can be attached to the surface of microbubbles or trapped in the bilayer of liposomes, or siRNA-loaded nanoparticles can be incorporated into ultrasound probes. For instance, epidermal growth factor receptor (EGFR)-directed siRNA (EGFR-siRNA) could be efficiently attached to microbubbles with around 7 mg siRNA per 109 microbubbles and safely protect siRNA from RNase digestion [166]. The EGFR-siRNA-loaded microbubbles reduced the EGFR expression of murine squamous carcinoma cells in vitro, and the ultrasound triggered destruction of microbubbles released EGFR-siRNA specifically in the tumor region to effectively delayed tumor growth, while the tumor volume was monitored by ultrasound.
However, microbubbles are limited to vascular compartment with poor tumor tissue penetration because of its large size and relatively poor stability. Therefore, ultrasound probes with much smaller size, such as nanobubbles, nanoparticles, and nanoscale liposomes, with better tumor tissue penetration properties have been fabricated for ultrasound diagnosis and ultrasound-mediated siRNA delivery with better tumor accumulation [157,167]. For instance, the ultrasound-sensitive siRNA nanobubbles made from positively charged liposomes with gas core and decorated with negatively charged siRNAs on the surface could effectively accumulate in the tumor tissues through EPR effect, demonstrating high potency for tumor imaging and targeted delivery of siRNA for RNAi therapy [168].Moreover, high cellular affinity ligands have been introduced to the surface of ultrasound nanoprobes, such as aptamer-decorated nanobubbles have been developed to specifically target the CCRF-CEM cells (T-cells, human acute lymphoblastic leukemia) for ultrasound imaging [159]. Other bioactive compounds, such as anticancer drugs and plasmid DNA, could also be co-loaded into probes for ultrasound imaging and combination therapy of tumors [160].
Multimodality imaging: Although each imaging modality has its unique advantages, it is also endowed with its intrinsic limitations making it difficult to obtain accurate and reliable information on all aspects of structure and function about the target organs by a single imagingmodality [169]. Table 2 summarizes some general features of classical imaging modalities including optical imaging, radionuclide imaging, MRI, and ultrasound imaging. To cope with the shortcomings of each modality, multimodality imaging combines different imaging techniques and imaging probes and can provide some complementary information about RNAi therapeutics. However, in context of developing multimodality probes, it should be noted that challenges are involved especially for applications in living subjects. Since the multimodality probes are primarily nanoparticle based, such as organic dye-labeled iron oxides, gadolinium chelates functionalized QDs, magnetic microbubbles, radiolabeled C-dots, etc. The major problems may be associated with insufficient concentration of probes at target sites due to the undesired uptake by mononuclear phagocyte system (MPS). Other concerns include slow clearance time, long-term retention in tissues and organs, as well as the long-term toxicity. These issues are highly related to the physicochemical properties (e.g., chemical component, size distribution, final hydrodynamic diameter, shape, surface charge) of nanoprobes [170]. In addition, different modalities differ in their imaging sensitivity by large magnitude [171]. Thus, the combination of two different probes needs to be carefully designed with proper ratio [172]. Therefore, in order to reach the full potential of multimodality imaging, the participations of multidisciplinary scientists with solid background in nanotechnology, material science, pharmacology, pharmaceutical chemistry, clinical medicine, biomedical engineering, and instrumental techniques are essential in the early development stages [173,174].
| Table 2: Features of different modalities for structural, functional, and molecular imaging of RNAi. | ||||||||
| Modality | Penetration depth | Sensitivity (Mol/L) |
Spatial resolution |
Physical medium |
Imaging probes and amount of use | Advantages | Disadvantages | |
| Optical FI |
< 1 cm | ~10−9–10−12 | 2–3 mm | Visible or NIR light |
Organic dyes; QDs; C-dots; UCNPs |
Microgram–milligram | High sensitivity; real-time imaging; nonionizing radiation; relatively inexpensive; short acquisition time; multiplexing capability |
Low spatial resolution at greater depth; limited penetration depth; autofluorescence |
| Optical BLI |
< 2 cm | ~10−15–10−17 | 3–5 mm | Visible light | GFP; RFP | Microgram–milligram | High sensitivity; real-time imaging; no ionizing radiation; relatively inexpensive; short acquisition time; user friendly; multiplexing capability |
Low spatial resolution at greater depth; Limited penetration depth; genetic reporter systems required |
| PET | Limitless | ~10−11–10−12 | 1–2 mm | High-nergy γ-rays |
Radiolabeled tracers |
Nanograms | Excellent sensitivity; limitless penetration depth; quantitative data |
High cost of cyclotron; Ionizing radiation; limited spatial resolution |
| SPECT | Limitless | 10−10–10−11 | 1–2 mm | Low-energy γ-rays |
Radiolabeled tracers |
Nanogram | Excellent sensitivity; limitless penetration depth; no need of cyclotron; multiplexing capability |
Relatively expensive; ionizing radiation; limited spatial resolution; semi-quantitative |
| MRI | Limitless | 10−3–10−5 | ~1 mm | Radio wav | Gadolinium chelates; Iron oxides; Other magnetic nanoparticles |
Microgram–milligram | Excellent spatial resolution; limitless penetration depth; quantitative data; no ionizing radiation |
Relatively expensive; relatively low sensitivity and poor contrast; long acquisition time |
| US | Millimeters–centimeters (frequency dependent) |
— | 0.01–2 mm |
Ultrasound waves |
Microbubbles | Microgram–milligram | Relatively inexpensive; no ionizing radiation; quantitative data; no ionizing radiation; short acquisition time; high sensitivity with microbubbles | Limited penetration depth; poor low contrast; strong boundary effect; limited to imaging soft tissue only |
| BLI: Bioluminescence Imaging; FI: Fluorescence Imaging; US: Ultrasound; NIR: Near-Infrared Fluorescence; GFP: Green Fluorescent Protein; RFP: Red Fluorescent Protein; QDs: Quantumdots; C-dots: Carbon nanodots; UCNPs: Upconvertion Nanoparticles. | ||||||||
A typical example of a multimodality probe-siRNA delivery system for in vivo imaging of RNAi has been reported by Medarova, et al. [175]. In this study, a dual-purpose probe was developed, which was composed of iron oxide core and Cy5.5 dye on the surface. The probe was further modified with cell membrane translocation peptides to facilitate intracellular delivery of siRNA (Figure 10a). The successful delivery of GFP siRNA duplex to tumors was assessed byMRI and optical imaging of tumor-bearing animals after intravenous injection of the probes (Figure 10 b,c). In another study, both commercial MRI contrast agents (Magnevist/Feridex) and Alexa-647 dye-labeled siRNA for targeting cyclooxygenase-2 (COX-2), an important therapeutic target in cancer, were encapsulated in PEGylated polycationic liposomes. The liposomes were used to assess the delivery and silencing effects of siRNA in vivo [176]. It was found that Feridex-loaded liposomes demonstrated better performance than that of Magnevist, which was further tested in vivo. Both MRI and optical imaging confirmed successful delivery of siRNA to MDA-MB-231 tumor.
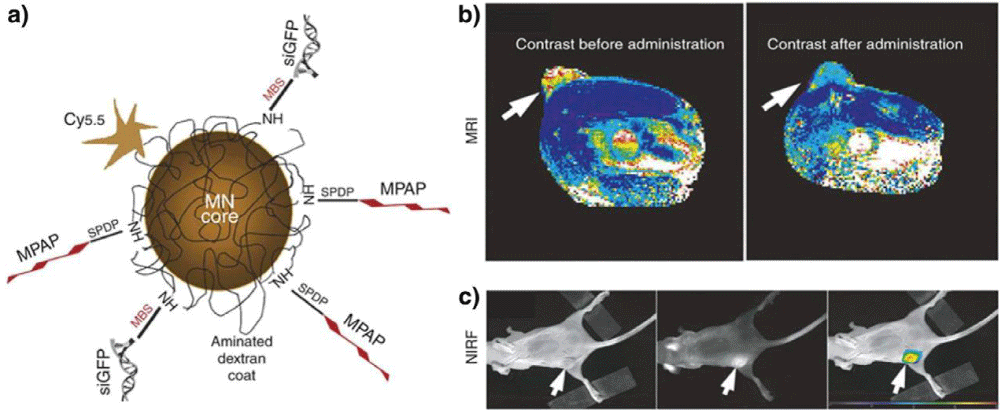
Figure 10: Multimodality in vivo imaging of RNAi by using MRI and optical imaging. (a) Schematic illustration of the multifunctional nanocarriers with core of magnetic nanoparticles, and surface conjugated with Cy5.5, GFP siRNA (siGFP), and membrane translocation peptides (MPAP). (b) in vivo MR imaging of mice bearing with subcutaneous LS174T human colorectal adenocarcinoma tumors (arrows) before and after the administration of the multifunctional nanocarriers, indicating significant drop of T2-weighted contrast enhancement in tumor regions after injection of the contrast agents. (c) A high-intensity NIRF signal in the tumor confirmed the successful delivery of the nanocarriers, while the left mouse with white light, middle one with NIRF, and right one with color-coded overlay. Adapted with permission from [175].
Recently, PET/CT combined with BLI was also employed to monitor the whole-body bio distribution of RNAi therapeutics and assess their silencing effects of the expression of luciferase in vivo [177]. The nanoparticles were prepared with cyclodextrin-containing polycations and anti-Luc siRNAs with their 5′-end conjugated with 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraaceticacid (DOTA) for 64Cu labeling. Micro-PET/CT was carried out to determine the distribution and tumor accumulation of siRNA-containing nanoparticles. No obvious difference in distribution between the targeted nanoparticles and nontargeted ones was observed. Meanwhile, the BLI revealed that the targeted nanoparticles had better RNAi effects 1 day after injection, demonstrating the importance of multimodal imaging. Thus, the combination of PET/CT and BLI is important to simultaneously monitor both the gene delivery and silencing effects of RNAi, which is critical for the design of RNAi therapeutics for clinical translation.
Technologies on theranostic nanomedicines
Theranostic nanomedicines contain both a diagnostic agent and one or more therapeutic drugs within one integrated system, enabling noninvasive diagnosis, therapy, and real-timemonitoring of the therapeutic response at the same time [178–181]. Among various imaging techniques, computed tomography (CT) is one of the most commonly used non-invasive clinical imaging modalities because of its wide availability, high spatial resolution, unlimited depth, and accurate anatomical information with reconstructed three dimensional imaging [182–184]. Iodixanol (Visipaque) is a small iodinated molecule, clinically used as a CT contrast agent that has a low osmolality and great tolerability [185]. However, like all low molecular weight iodinated CT contrast agents, iodixanol has drawbacks like non-specific distribution and rapid renal clearance following i.v. injection [186]. In recent years, nanosized CT contrast agents have attracted great interest as they have several advantages over small molecular contrast agents such as prolonged circulation time, site-specific accumulation and use for theranostics [187–190]. Some recent work showed systems with great promise of nanosized CT contrast agents such as iodinated hyaluronic acid oligomer-based nano-assembled systems, theranostic self-assembly structures of gold nanoparticles, and multifunctional dendrimerentrapped gold nanoparticles for simultaneous tumor imaging and therapy [191–193].
Among various types of nanoscale drug delivery systems, nanogels have attracted increasing attention since they have a large surface area for multivalent bioconjugation and a cross linked three-dimensional network structure that offers great colloidal stability [194–196]. To achieve rapid release of the payload at the target site, pH, redox potential, and enzyme-responsive nanogels have been designed [197–204]. Nanogels based on hyaluronic acid (HA) have recently appeared as a unique system because HA is a hydrophilic natural material with excellent biocompatibility and intrinsic targeting ability toward CD44-overexpressing tumor cells [203,205–208]. HA nanoparticles have been used for efficient delivery of chemotherapeutics, proteins as well as siRNA in vitro and in vivo [209–212].
We report on bioresponsive and fluorescent hyaluronic acid-iodixanol nanogels (HAI-NGs) for targeted CT imaging and chemotherapy of MCF-7 human breast tumor (Scheme 1). HAI-NGs were obtained from hyaluronic acid-cystamine-tetrazole (HA-Cys-Tet) and reductively degradable polyiodixanol-methacrylate (SS-PI-MA) via nanoprecipitation and a photo-click crosslinking reaction. HAI-NGs were designed with the following unique features: i) both HA and iodixanol have excellent biocompatibility and are currently used in the clinic; ii) the “tetrazole-ene” photo-click crosslinking reaction is highly selective, which prevents cross-reaction with most drugs and furthermore endows nanogels with bright green fluorescence [213,214]; iii) HA can actively target CD44 receptors which are overexpressed on various malignant tumor cells and stem cells [215–218]; iv) HAI-NGs can be used for targeted CT imaging in vivo; and v) the reduction-sensitivity of HAI-NGs allows fast intracellular release of payloads like PTX to achieve efficient and targeted chemotherapy. Tetrazole (Tet) and cystamine diisocyanate (CDI) were synthesized according to previous reports [213,219]. Herein, the stability of HAI-NGs and the reduction-triggered PTX release from PTX loaded HAI-NGs were investigated. Furthermore, the targetability of HAI-NGs and antitumor activity of PTX loaded HAI-NGs toward MCF-7 cells, the pharmacokinetics and bio distribution, NIR and CT imaging, as well as therapeutic effects in MCF-7 human breast tumor xenografts in mice were evaluated.
Scheme. 1 Illustration of bioresponsive and fluorescent hyaluronic acid-iodixanol nanogels for targeted X-ray computed tomography imaging and chemotherapy of breast tumors. (a) PTX-loaded HAI-NGs are prepared via nanoprecipitation followed by crosslinking via UV irradiation; (b) PTX-loaded HAI-NGs actively target and accumulate at MCF-7 tumors, resulting in enhanced CT contrast and targeted therapy; (c) PTX-loaded HAI-NGs are selectively internalized into the MCF-7 breast tumor cells via CD44 receptor-mediated endocytosis, nanogels are decrosslinked and disassembled in response to GSH in the cytosol, and PTX is quickly released into the cells.
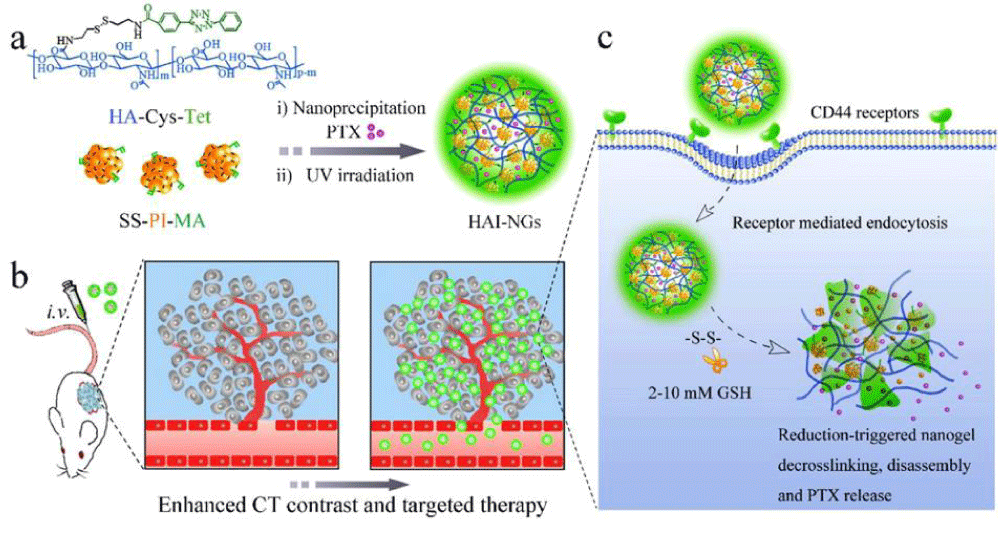
Scheme 1: Illustration of bioresponsive and fluorescent hyaluronic acid-iodixanol nanogels for targeted X-ray computed tomography imaging and chemotherapy of breast tumors. (a) PTX-loaded HAI-NGs are prepared via nanoprecipitation followed by crosslinking via UV irradiation; (b) PTX-loaded HAI-NGs actively target and accumulate at MCF-7 tumors, resulting in enhanced CT contrast and targeted therapy; (c) PTX-loaded HAI-NGs are selectively internalized into the MCF-7 breast tumor cells via CD44 receptor-mediated endocytosis, nanogels are decross linked and disassembled in response to GSH in the cytosol, and PTX is quickly released into the cells.
Preparation of nanogels and triggered drug release: Hyaluronic acid-iodixanol nanogels (HAI-NGs) were readily obtained via nanoprecipitaion and photo-click crosslinking reaction from HACys-Tet and SS-PI-MA. Figure 11A shows that HAI-NGs had a small size of about 90 nm with a low polydispersity (PDI) of 0.11. TEM confirmed that HAI-NGs had a homogenous size distribution and spherical morphology (Figure 11B). Notably, HAI-NGs emitted bright green fluorescence under UV light (Figure 11C inset), which derives from pyrazoline cycloadducts produced by the “tetrazole-alkene” photo-click reaction [213,220]. Fluorescence spectroscopy displayed that HAI-NGs had a strong emission at ca. 485 nm (Figure 11C). The strong fluorescence of HAI-NGs can be used to monitor their in vitro and in vivo fate. HAI-NGs displayed excellent stability against extensive dilution as well as 10% serum. However, in the presence of 10 mM glutathione (GSH), HAI-NGs rapidly swelled and agglomerated, supporting their fast redoxresponsivity (Figure 11D).
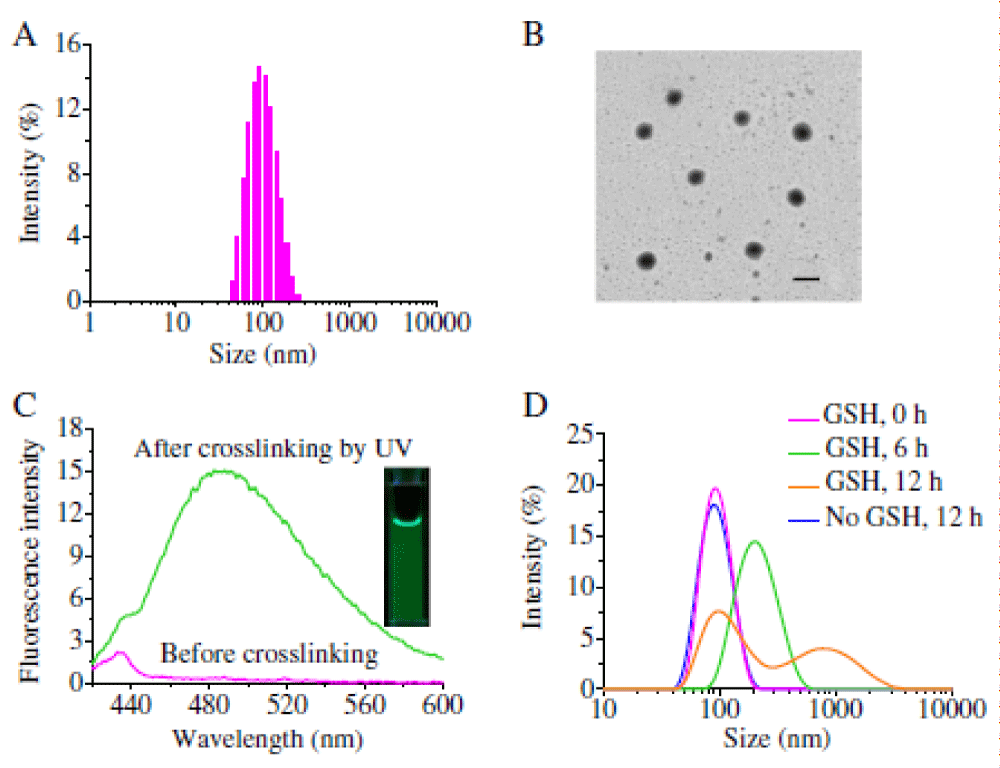
Figure 11: (A) Intensity size distribution of HAI-NGs determined by DLS. (B) TEM image of HAI-NGs (scale bar: 100 nm). (C) Fluorescent spectrum of HAI-NGs before and after crosslinking by UV irradiation. The insert shows a photograph of HAI-NGs under UV light. (D) Triggered destabilization of HAI-NGs in 10 mM GSH.
In contrast, nearly complete PTX release was observed in the presence of 10 mM GSH under otherwise the same conditions, probably due to GSH triggered disulfide bond cleavage and de-crosslinking of the nanogels, corroborating that drug release can be accelerated in an intracellular reductive environment. Nanogels typically have a low loading and fast leakage of small molecule drugs [221]. Paclitaxel (PTX) could be easily loaded into HAI-NGs during nanoprecipitation. The high PTX loading and inhibited drug leakage of HAI-NGs is likely due to existence of strong π-π interactions between PTX and pyrazoline groups and iodixanol moieties in the nanogels [222].
Cellular uptake and cytotoxicity of PTX-loaded HAI-NGs: Given their strong fluorescence, the cellular uptake of HAI-NGs into CD44 receptor overexpressing MCF-7 breast cancer cells could be conveniently traced by confocal laser scanning microscopy (CLSM). Notably, nanogel fluorescence was clearly observed in MCF-7 cells after 1 h incubation and the fluorescence became stronger at a prolonged incubation time of 2 or 4 h (Figure 12) [223]. The cellular uptake of nanogels was greatly inhibited and only weak nanogel fluorescence was discerned in the cellmembrane of MCF-7 cells pre-incubated for 4 h with free HA, demonstrating that HAI-NGs are internalized byMCF-7 cells via a receptor-mediated mechanism. Here it has been also selected L929 murine fibroblastic cells with a low expression of CD44 as negative controls, the fluorescence intensity of HAI-NGs in MCF-7 cells was much stronger than that in L929 cells, again proving the cellular uptake of HAI-NGs via a CD44-mediated mechanism.
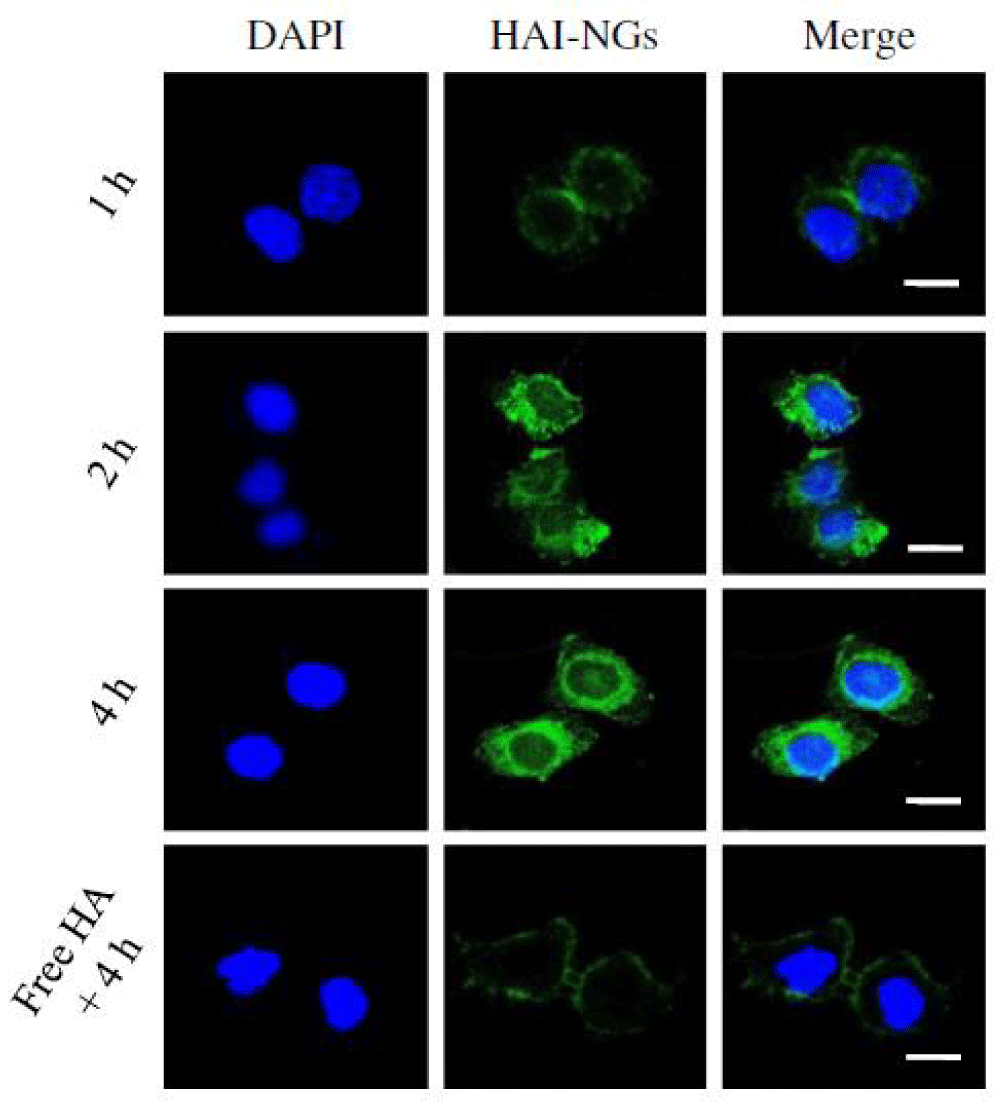
Figure 12: CLSM images ofMCF-7 cells following 1 h, 2 h and 4 h incubation with HAI-NGs. Cells pre-treated with free HA (5 mg/mL) for 4 h before adding nanogels were used as a control. The scale bars correspond to 20 μm in all the images.
MTT assays showed that blank HAI-NGs were practically non-toxic to MCF-7 cells (N93% cell viability) even at a high nanogel concentration of 1 mg/mL (Figure 13A), indicating that HAI-NGs possess excellent biocompatibility. In contrast, PTX-loaded HAI-NGs exhibited significant and dose dependent cytotoxicity against MCF-7 cells (Figure 13B). The half-maximal inhibitory concentration (IC50) of PTX-loaded HAI-NGs was determined to be 0.52 μg/mL, comparable to that of free PTX (0.35μg/mL), corroborating their efficient cellular internalization and rapid intracellular PTX release. The pre-treatment of MCF-7 cells with free HA for 4 h largely reduced the cytotoxic effect of PTX-loaded HAI-NGs, in line with the above CLSM observations that cellular uptake is inhibited by free HA.
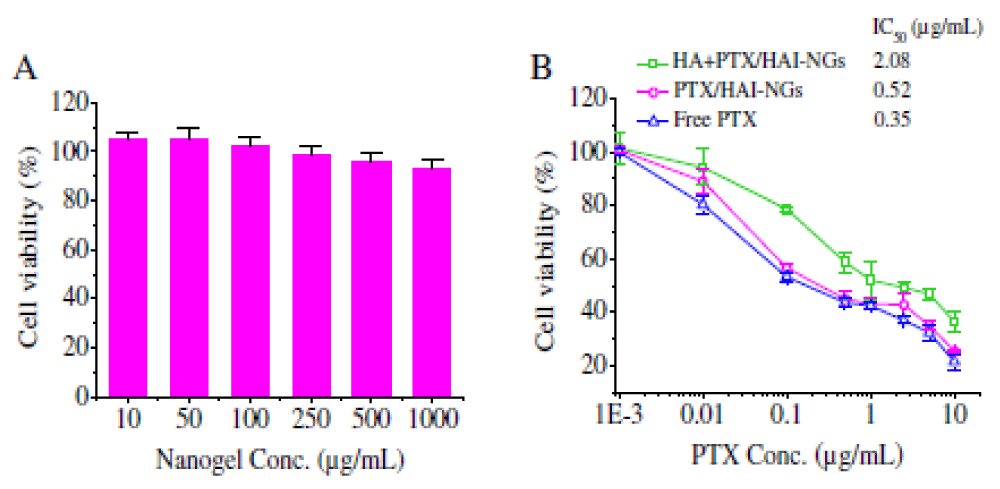
Figure 13: MTT assays of blank HAI-NGs and PTX/HAI-NGs inMCF-7 cells. (A) Viability of MCF-7 cells following 48 h incubation with blank HAI-NGs; and (B) in vitro antitumor activity of PTX/ HAI-NGs againstMCF-7 cells. The cells were incubated with PTX/HAI-NGs for 4 h, the medium was removed and replenished with fresh culture medium, and the cells were cultured for an additional 44 h. The inhibition experiment was performed by pre-treating cells for 4 h with free HA (5 mg/mL) prior to incubation with PTX/HAI-NGs. Data are presented as mean ± SD (n = 4).
in vivo pharmacokinetics, near infrared imaging and bio distribution of nanogels: To investigate the in vivo pharmacokinetics, PTX-loaded HAI-NGs were i.v. injected into BALB/c nude mice at 5 mg PTX/kg and the plasma levels of PTX at different time points were determined by HPLC. Figure 14A displays that PTX-loaded HAI-NGs had a prolonged circulation time with an elimination half-life of 3.3 h. [223] indicating that nanogels are stable in the circulation and drug leakage is low as a result of strong π-π interactions between PTX and pyrazoline group and iodixanol moieties in the nanogels [222]. In comparison, free PTX was rapidly eliminated from the blood circulation, with an extremely short half-life time of 0.35 h.
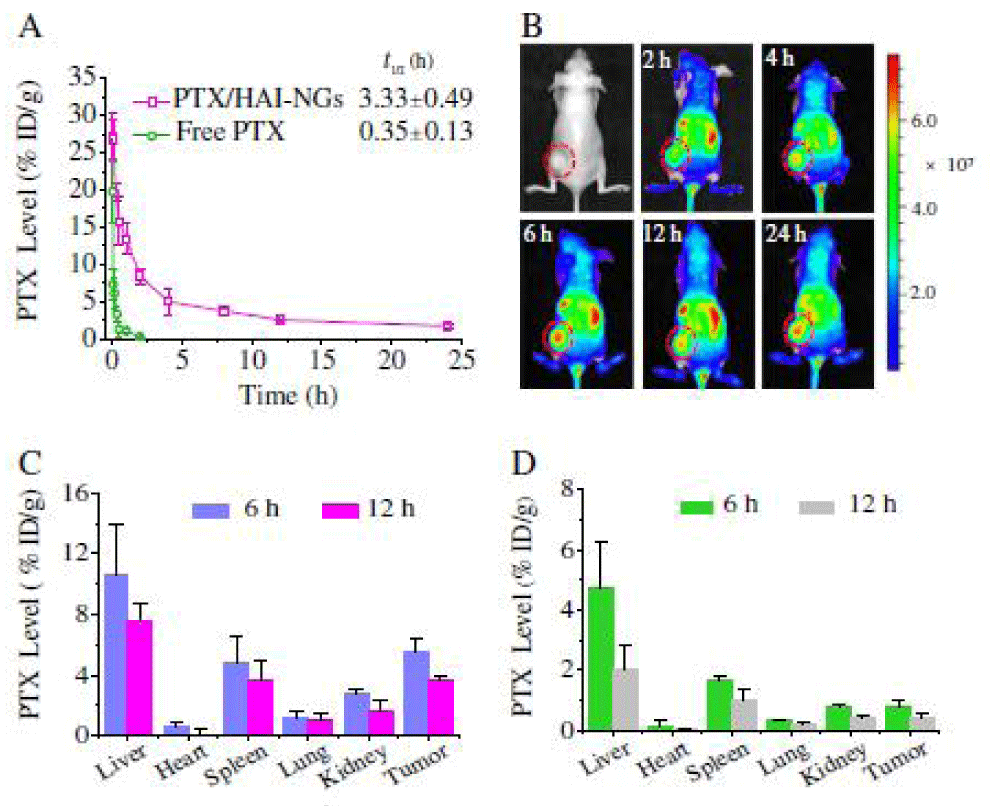
Figure 14: in vivo pharmacokinetics, NIR imaging and bio distribution studies. (A) in vivo pharmacokinetics of PTX/HAI-NGs and free PTX in mice. PTX level is expressed as % injected dose per gram of tissue (%ID/g). Data are presented as mean ± SD (n = 3); (B) in vivo fluorescence images of MCF-7 human breast tumor bearing nude mice at different time points following tail vein injection of Cy5-loaded nanogels. Circled areas indicate the locations of the tumor. PTX level, expressed as % injected dose per gram of tissue (% ID/g), in the tumor and different organs after 6 or 12 h post injection of PTX/HAI-NGs (C) or free PTX (D). Data are presented as mean ± SD (n = 3).
To visualize their tumor accumulation in vivo, a near infrared dye Cy5was loaded into HAI-NGs, and the serumCy5 release was evaluated. It has been found that Cy5 release from HAI-NGs was slow and within 24 h only 4.1% was released. Therefore it is envisaged that Cy5 loaded HAI-NGs are also relatively stable in the circulation. Figure 14B shows real-time images of Cy5-loaded HAI-NGs in MCF-7 tumor-bearing mice. Notably, tumor accumulation of nanogels was clearly observed at 2 h post injection and reached a maximum at 6 h. This high tumor targeting efficiency of nanogels is likely due to their small size, high stability, and active targeting effect. Interestingly, the fluorescence at the tumor site at 24 h again became stronger compared with that at 12 h, probably because the Cy5 fluorescence is partly self-quenched when loaded into the HAI-NGs due to the homo Förster resonance energy transfer (homo-FRET) effect [224]. When Cy5 molecules are partly released from the disassembled HAI-NGs in the reductive environment, the fluorescence of Cy5 in the tumor area may increase again. It is also noticed that besides in the tumor site, strong fluorescence was also observed in the liver and spleen, probably because after i.v. injection, nanogels are also captured by the RES system (mainly liver and spleen). As CD44 receptors are also expressed on liver and spleen cells, uptake of part of the nanogels by these organs may be inevitable”.
To profile the in vivo bio distribution of PTX-loaded HAI-NGs inMCF-7 tumor bearing mice, PTX levels in the tumor and different organs at 6 and 12 h post injection were quantitatively determined by HPLC. Notably, PTX-loaded nanogels exhibited a high tumor accumulation with 5.5%ID/g at 6 h (Figure 14C). The tumor PTX accumulation remained high (3.6% ID/g) at 12 h post injection. In comparison, free PTX displayed 7-and 15-fold lower tumor accumulation than PTX-loaded HAI-NGs at 6 and 12 h post injection, respectively (Figure 14D versus Figure 14C). The PTX level expressed in %ID for the tissues after injection with PTX/HAI-NGs also showed a relatively high retention of PTX in tumor tissue besides the high level in liver tissue and lower levels in the spleen. The levels after the administration of free PTX were investigated [223]. The pharmacokinetics, NIR imaging and biodistribution studies all point out that PTX-loaded HAI-NGs have a prolonged circulation time and significantly enhance PTX accumulation in the MCF-7 tumor.
Enhanced CT imaging by HAI-NGs: HAI-NGs can be used for CT imaging due to the presence of a high content of iodixanol. Figure 15A shows clearly that HAI-NGs effectively enhanced the CT contrast in vitro. The corresponding Hounsfield units (HU) values exhibited a linear correlation with HAI-NGs concentrations, suggesting that HAI-NGs can be used for quantitative CT studies. It has been then evaluated the application of HAI-NGs for in vivo CT diagnosis. Interestingly, 5 min after intratumoral (i.t.) injection of 50 μL of HAI-NGs at an HAI-NGs concentration of 15 mg/mL (i.e. 60 mg iodine equiv./kg) into MCF-7 tumor bearing nude mice, remarkably enhanced contrast was observed at the tumor site in the three-dimensional reconstructed images, with amarked increase of HU value from 37.2 to 182.8 (Figure 15B). It has been further investigated whether HAI-NGs can be applied for targeted CT imaging of CD44 overexpressed tumors.
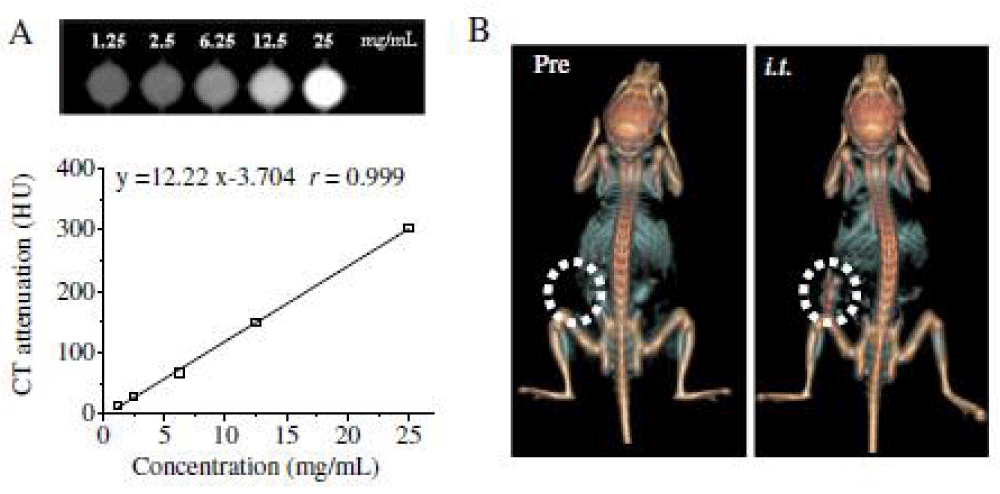
Figure 15: (A) HU measurements of HAI-NGs. The plot showed a linear correlation of CT calculated solution attenuation measured in Hounsfield units (HU) to HAI-NGs at varying concentrations from 1.25 to 25 mg/mL. (B) Three-dimensional reconstructed CT images of the MCF-7 tumor bearing mice following intratumoral (i.t.) injection of 50 μL of HAI-NGs at a concentration of 15 mg/mL (60 mg iodine equiv./kg). Circled areas indicate the location of the tumor.
The results showed that enhanced contrast was discerned in the MCF-7 tumor from both axial and coronal CT images, with HU values increasing from 37.0 to 82.6, at 7 h following intravenous injection of HAI-NGs into MCF-7 tumor bearing nude mice (Figure 16). The enhanced tumor contrast further confirms that HAI-NGs can target to and accumulate in the MCF-7 tumor. Notably, the high contrast signal at the tumor site lasted for a long time, which is advantageous for clinical diagnosis. In sharp contrast, little enhancement of HU value was observed at the tumor site for iodixanol (small molecule contrast agent) at the same iodine dose. Iodixanol was rapidly cleared from the body to the bladder. It is clear, therefore, that HAINGs are superior to iodixanol in targeted CT imaging of CD44 positive tumors.
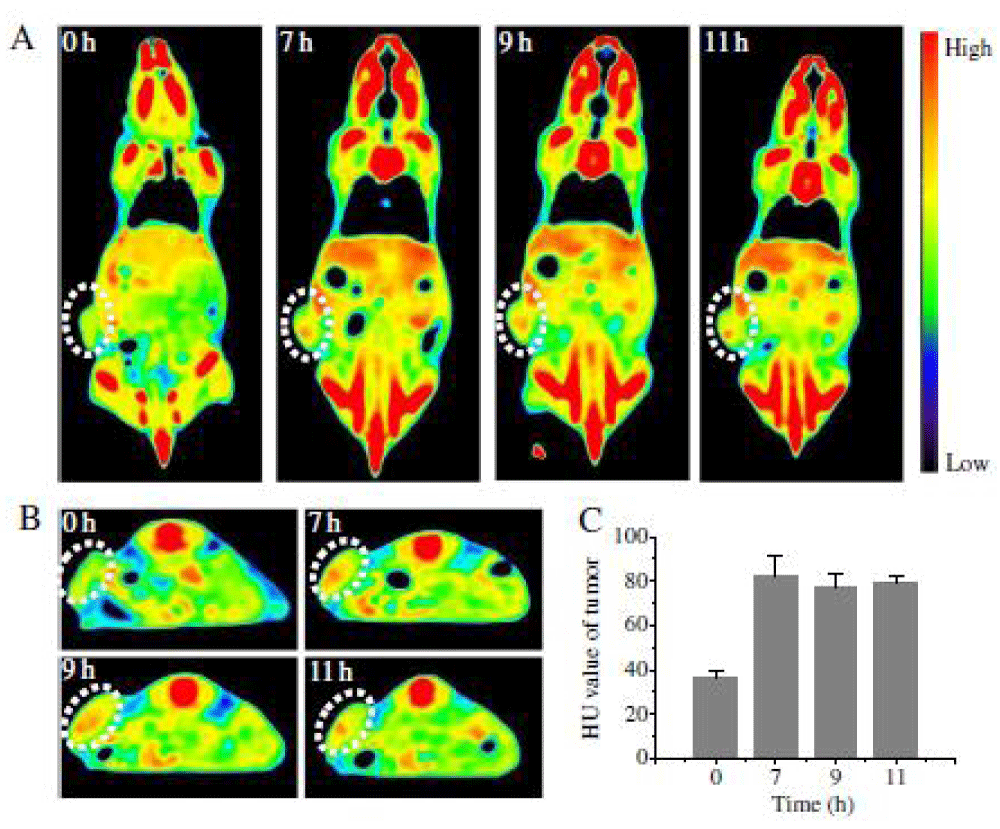
Figure 16: Axial CT images (A), coronal CT images (B) and HU value (C) of MCF-7 tumor bearing mice at 0 h, 7 h, 9 h and 11 h following tail vein injection of HAI-NGs at a concentration of 15 mg/mL (60 mg iodine equiv./kg). Circled areas indicate the position of the tumor.
in vivo tumor penetration and therapeutic efficacy of PTX-loaded HAINGs
In the process of tumor-targeted drug delivery, a series of biological barriers may influence the final therapeutic efficacy, among which are interstitial hindrance and tumor penetration [225,226]. The strong intrinsic fluorescence of HAI-NGs was utilized to track their distribution. The blood vessels and cell nuclei were stained by CD31 antibody and DAPI, respectively. Figure 17 reveals that HAI-NGs were located in the blood vessels at 2 h post i.v. injection. At a prolonged time of 6 h post injection, HAI-NGs extravagated from blood vessels to the interstitial space, displaying green fluorescence throughout the whole tumor. At 12 h post injection, HAI-NGs penetrated further deep into the tumor and were actively endocytosed by tumor cells, presenting bright green fluorescence around the cell nuclei. The above phenomena suggest that the HAI-NGs possess good tumor penetration ability.
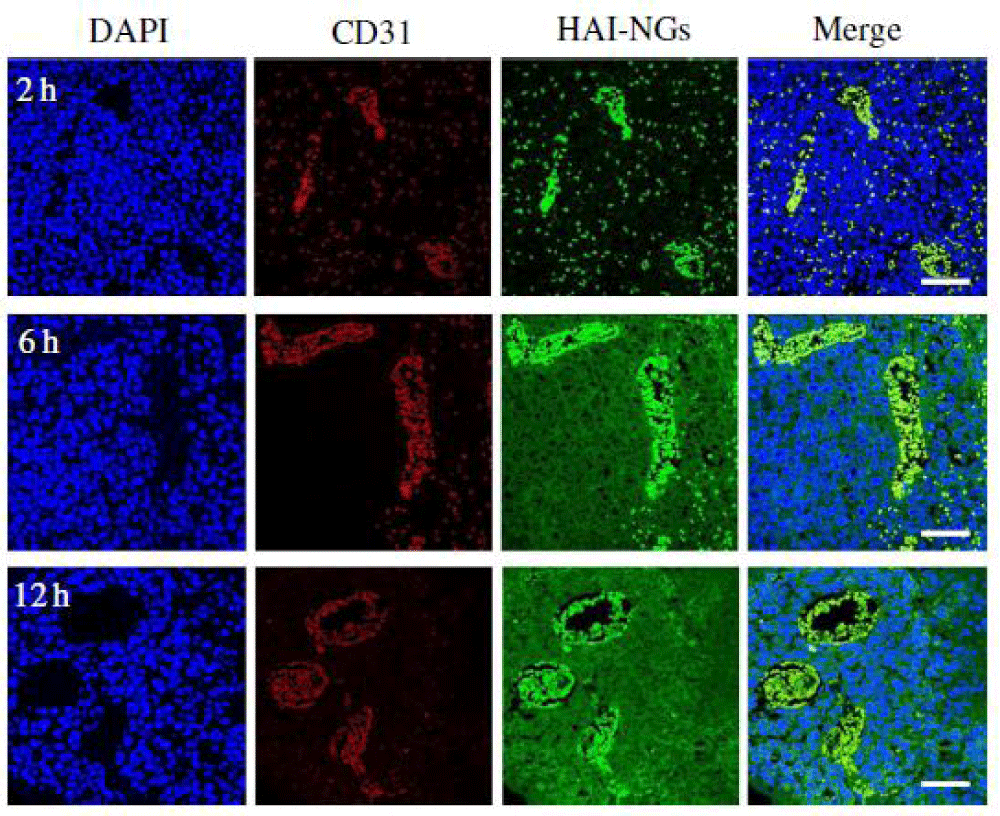
Figure 17: Tumor penetration of HAI-NGs observed by confocal microscopy. Tumor sections were obtained fromMCF-7 tumor bearing mice following 2, 6 and 12 h tail vein injection of HAINGs (10 mg/mL). The nuclei were stained with DAPI (blue) and blood vessels were stained with CD31 (red). HAI-NGs have an intrinsic green fluorescence. The scale bar represents 50 μm.
The antitumor efficacy of PTX-loaded HAI-NGs was evaluated in MCF-7 tumor bearing mice at a dose of 5 mg PTX equiv./kg. The results [223] showed that PTX-loaded HAI-NGs exhibited effective inhibition of tumor growth, which was significantly better than that of free PTX (Figure 18A). The photographs of tumor blocks excised on day 24 further confirmed that mice treated with PTX-loaded HAI-NGs had the smallest tumor size (Figure 18B). Both PTX-loaded HAI-NGs and free PTX caused no change of mice body weight (Figure 18C). Importantly, survival curves showed that PTX-loaded HAI-NGs effectively prolonged the survival time of the MCF-7 breast tumor bearing mice with all mice surviving over an experimental period of 65 d (Figure 18D). In contrast, mice treated with free PTX and PBS had a median survival time of 40 and 28 d, respectively. The histological analyses by H&E staining displayed that PTX-loaded HAI-NGs induced widespread necrosis of tumor tissue with little damage to the healthy organs including heart, liver and kidney (Figure 18E), supporting that PTX-loaded HAI-NGs cause low systemic side effects. In comparison, hepatocellular necrosis (red arrows) was observed for free PTX treated mice, similar to previous reports [227,228]. It is evident that PTX-loaded HAI-NGs have low systemic toxicity and mediate efficient and targeted delivery of PTX to human breast tumors in vivo, resulting in effective suppression of tumor growth and markedly prolonged survival time.
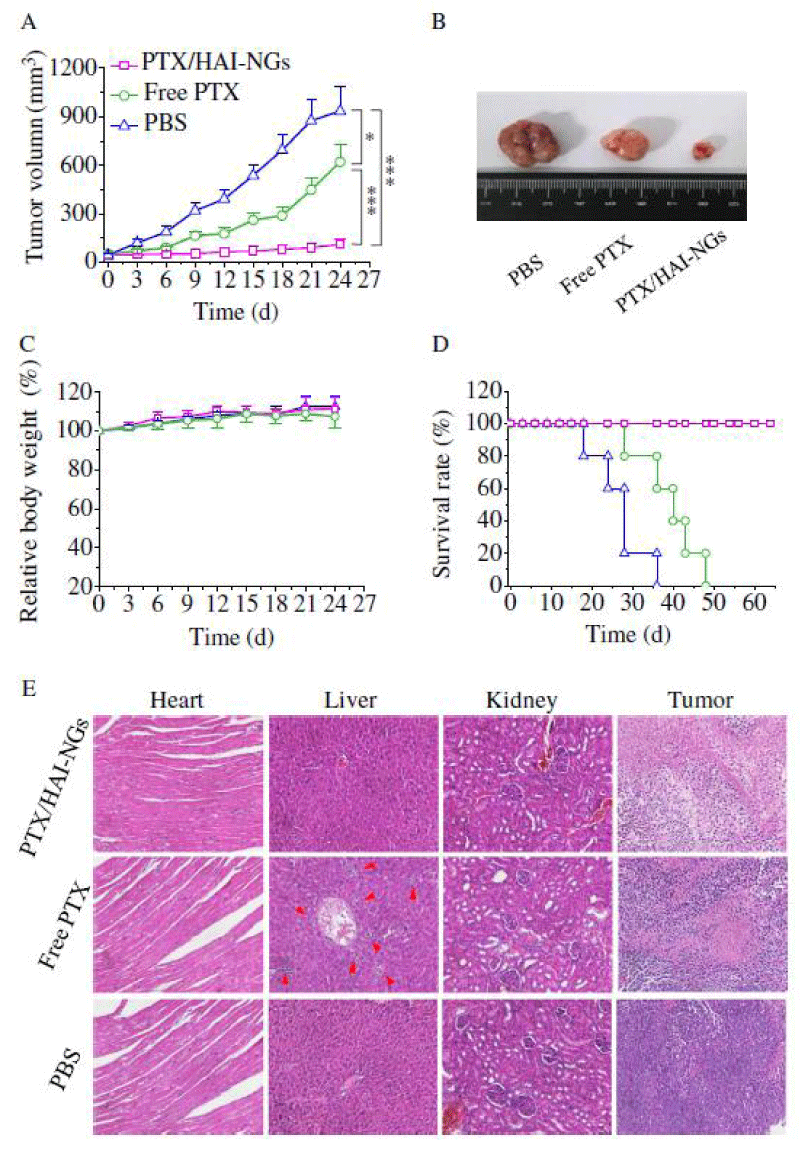
Figure 18: in vivo antitumor efficacy of PTX/HAI-NGs inMCF-7 tumor bearing nude mice. Free PTX and PBS were used as controls. The drug was given on day 0, 3, 6, 9, 12 (drug dosage: 5mg PTX equiv/kg). (A) Tumor volume changes in time. Data are presented as mean ± SD (n = 6). (B) Photographs of tumor blocks collected from different treatment groups on day 24. (C) Body weight changes of nude mice following different treatments within 24 days. (D) Survival rates of mice in different treatment groups within 60 d. Data are presented as means ± SD (n = 5). *p < 0.05, *** p < 0.001. (E) H&E stained heart, liver, kidney and tumor sections excised from MCF-7 human breast tumor-bearing mice following 24 d treatment with PTX/HAINGs, free PTX or PBS. The images were obtained by a Leica microscope at 200× magnification. Red arrows indicate hepatocellular necrosis.
This chapter summarized the application of various imaging modalities for qualitative and quantitative assessments of RNAi therapeutics to promote their applications in cancer therapy and translate them into clinical applications. Overall, a variety of RNAi therapeutics has been developed and some of them are under clinical evaluation. Diverse imaging techniques have been applied to study the mechanism of gene transfection, route of delivery, systemic distribution in the body, genesilencing effect, and therapeutic outcomes.
RNAi has demonstrated great potential for treating a wide range of diseases, especially for multidrug-resistant cancer treatment, which are difficult to be treated by conventional methods of chemotherapy and radiotherapy. Despite the positive progresses in translating some RNAi formulations into clinical trial for cancer therapy, several issues are required to bear in mind for effective translation of RNAi therapeutics from bench to bedside: (1) How to optimize the parameters/conditions to accumulate sufficient amount of RNAi therapeutics in tumor tissues and translocate them into cancer cells; (2) How to select the proper target to maximize the therapeutic effects of siRNA; and (3) How to develop new effective gene carriers with high stability during circulation as well as controlled release function of payload nucleic acids inside cancer cells with high gene transfection effects. Finally, the ideal RNAi formulations should satisfy the requirement of specific delivery of RNAi agents to tumors and efficiently transfer into cancer cells to silence target genes at high performance for tumor suppression, while avoiding potential toxicity, side effects, and off-target silencing.
The advances in noninvasive imaging techniques [229-234] provide new approaches to visualize and quantify RNAi in cancer therapy and further understand the mechanism of RNAi intracellular and in the body. So far, several imaging modalities, such as optical imaging, MRI, ultrasound, and SPECT/PET, have been applied to assess the delivery of siRNA, to determine the bio distribution, and to monitor the therapeutic effects, leading to a significant contribution to the progresses of RNAi therapeutics, especially in the preclinical studies [235,236]. It is very important to establish proper evaluation models/systems with the aid of molecular imaging in preclinical studies and translate the animal results as important references for clinical trial in human. As each imaging modality has its intrinsic advantages and limitations, there is a trend to combine different imaging modalities for better tracing the fate of siRNA, the delivery vehicles, and the therapeutic effects, which is expected to maximize the potential of cancer therapywith RNAi therapeutics and help overcome the barriers that block the road to clinical translation.
The rational design of molecular imaging probes is essential to accurately monitor the biological processes of RNAi therapeutics and fully realize their potency in cancer therapy. Molecular imaging probes for RNAi-based cancer therapeutics, consisting of a variety of functional nanomaterials including lipids, metals, carbons, polymers, and biologics-based nanoparticles, should be relatively safe for clinical use with low toxicity and biodegradability in the body. Further development of new imaging contrast agents, which can increase the signal intensity for more reliable image analysis with cancer cell specific targeting ability, would optimize the diagnostic selectivity and provide more detailed and accurate pathological information about the biological processes of RNAi therapeutics in cancer treatment. Furthermore, the technical advances in imaging devices would enable proper patient selection and rapid translation of RNAi therapeutics into clinic.
We have demonstrated for the first time that bioresponsive and fluorescent hyaluronic acid-iodixanol nanogels (HAI-NGs) mediate targeted X-ray computed tomography (CT) imaging and chemotherapy of MCF-7 human breast tumor in vivo. Notably, HAI-NGs have integrated multiple functions including excellent biocompatibility, bright green fluoresence, high stability, superior target ability to CD44 overexpressing cells and fast glutathione-responsive drug release. The in vivo studies clearly show that PTX-loaded HAI-NGs have a prolonged circulation time, high tumor accumulation and enhanced tumor penetration in MCF-7 breast tumor-bearing nude mice, resulting in effective tumor growth inhibition, markedly improved survival rate, and reduced systemic toxicity as compared to free PTX. Furthermore, HAI-NGs via either intratumoral or intravenous injection leads to significantly enhanced CT imaging of MCF-7 breast tumors in nude mice as compared to iodoxanol. HAI-NGs provide a highly versatile and targeted theranostic nanoplatform that elegantly combines CT imaging with targeted chemotherapy toward CD44 overexpressing tumors.
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, et al, Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998; 391: 806–811. PubMed: https://pubmed.ncbi.nlm.nih.gov/9486653/
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells, Nature. 2001; 411: 494–498. PubMed: https://pubmed.ncbi.nlm.nih.gov/11373684/
- Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2012—an update. J Gene Med. 2013; 15: 65–77. PubMed: https://pubmed.ncbi.nlm.nih.gov/23355455/
- Thi EP, Mire CE, Ursic-Bedoya R, Geisbert JB, Lee ACH, et al. Marburg virus infection in nonhuman primates: therapeutic treatment by lipid-encapsulated siRNA. Sci Transl Med. 2014; 6: 250ra116. PubMed: https://pubmed.ncbi.nlm.nih.gov/25143366/
- Kaiser PK, Symons RC, Shah SM, Quinlan EJ, Tabandeh H, et al. RNAi-based treatment for neovascular age-related macular degeneration by Sirna-027. Am J Ophthalmol. 2010; 150: 33–39. PubMed: https://pubmed.ncbi.nlm.nih.gov/20609706/
- O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010; 10: 111–122. PubMed: https://pubmed.ncbi.nlm.nih.gov/20098459/
- van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov. 2012; 11: 860–872. PubMed: https://pubmed.ncbi.nlm.nih.gov/23080337/
- Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013; 369: 819–829. PubMed: https://pubmed.ncbi.nlm.nih.gov/23984729/
- Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010; 464: 1067–1070. PubMed: https://pubmed.ncbi.nlm.nih.gov/20305636/
- Shen H, Sun T, Ferrari M. Nanovector delivery of siRNA for cancer therapy. Cancer Gene Ther. 2012; 19: 367–373. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3842228/
- Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013; 3: 406–417. PubMed: https://pubmed.ncbi.nlm.nih.gov/23358650/
- Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008; 5: 505–515. PubMed: https://pubmed.ncbi.nlm.nih.gov/18672949/
- Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2010; 8: 129–138. PubMed: https://pubmed.ncbi.nlm.nih.gov/19180106/
- Li W, Szoka FC, Jr. Lipid-based nanoparticles for nucleic acid delivery. Pharm Res. 2007; 24: 438–449. PubMed: https://pubmed.ncbi.nlm.nih.gov/17252188/
- Mintzer MA, Simanek EE. Nonviral vectors for gene delivery. Chem Rev. 2009; 109: 259–302. PubMed: https://pubmed.ncbi.nlm.nih.gov/19053809/
- Zumbuehl AA, Goldberg M, Leshchiner ES, Busini V, Hossain N, et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat Biotechnol. 2008; 26: 561–569. PubMed: https://pubmed.ncbi.nlm.nih.gov/18438401/
- Semple SC, Akinc A, Chen J, Sandhu AP, Mui BL, et al. Rational design of cationic lipids for siRNA delivery. Nat Biotechnol. 2010; 28: 172–176. PubMed: https://pubmed.ncbi.nlm.nih.gov/20081866/
- Lee CC, MacKay JA, Frechet JM, Szoka FC. Designing dendrimers for biological applications. Nat Biotechnol. 2005; 23: 1517–1526. PubMed: https://pubmed.ncbi.nlm.nih.gov/16333296/
- Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov. 2005; 4: 581–593. PubMed: https://pubmed.ncbi.nlm.nih.gov/16052241/
- Thomas M, Klibanov AM. Non-viral gene therapy: polycation-mediated DNA delivery. Appl Microbiol Biotechnol. 2003; 62: 27–34. PubMed: https://pubmed.ncbi.nlm.nih.gov/12719940/
- Mo RH, Zaro JL, Shen WC. Comparison of cationic and amphipathic cell penetrating peptides for siRNA delivery and efficacy. Mol Pharm. 2012; 9: 299–309. PubMed: https://pubmed.ncbi.nlm.nih.gov/22171592/
- Martin ME, Rice KG. Peptide-guided gene delivery. AAPS J. 2007; 9: E18–E29. PubMed: https://pubmed.ncbi.nlm.nih.gov/17408236/
- Dassie JP, Liu XY, Thomas GS, Whitaker RM, Thiel KW, et al, Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMAexpressing tumors. Nat Biotechnol. 2009; 27: 839–849. PubMed: https://pubmed.ncbi.nlm.nih.gov/19701187/
- Yao YD, Sun TM, Huang SY, Dou S, Lin L, et al. Targeted delivery of PLK1-siRNA by ScFv suppresses Her2+ breast cancer growth and metastasis. Sci Transl Med. 2012; 4: 130ra148. PubMed: https://pubmed.ncbi.nlm.nih.gov/22517885/
- Choi KY, Silvestre OF, Huang X, Hida N, Liu G, et al. A nanoparticle formula for delivering siRNA or miRNAs to tumor cells in cell culture and in vivo. Nat Protoc. 2014; 9: 1900–1915. PubMed: https://pubmed.ncbi.nlm.nih.gov/25033207/
- Choi KY, Silvestre OF, Huang X, Min KH, Howard GP, et al. Versatile RNA interference nanoplatform for systemic delivery of RNAs. ACS Nano. 2014; 8: 4559–4570. PubMed: https://pubmed.ncbi.nlm.nih.gov/24779637/
- Liu G, Choi KY, Bhirde A, Swierczewska M, Yin J, et al. Sticky nanoparticles: a platform for siRNA delivery by a bis(zinc(II) dipicolylamine)-functionalized, self-assembled nanoconjugate. Angew Chem Int. 2012; 51: 445–449. PubMed: https://pubmed.ncbi.nlm.nih.gov/22110006/
- Sokolova V, Epple M. Inorganic nanoparticles as carriers of nucleic acids into cells. Angew Chem Int. Ed Engl. 2008; 47: 1382–1395. PubMed: https://pubmed.ncbi.nlm.nih.gov/18098258/
- Afonin KA, Viard M, Koyfman AY, Martins AN, Kasprzak WK, et al. Multifunctional RNA nanoparticles. Nano Lett. 2014; 14: 5662–5671. PubMed: https://pubmed.ncbi.nlm.nih.gov/25267559/
- Cui DX, Zhang CL, Liu B, Shu Y, Du T, et al. Regression of gastric cancer by systemic injection of RNA nanoparticles carrying both ligand and siRNA. Sci Rep. 2015; 5: e10726. PubMed: https://pubmed.ncbi.nlm.nih.gov/26137913/
- Lee H, Lytton-Jean AKR, Chen Y, Love KT, Park AI, et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat Nanotechnol. 2012; 7: 389–393. PubMed: https://pubmed.ncbi.nlm.nih.gov/22659608/
- Rychahou P, Haque F, Shu Y, Zaytseva Y, Weiss HL, et al. Delivery of RNA nanoparticles into colorectal cancer metastases following systemic administration. ACS Nano. 2015; 9: 1108–1116. PubMed: https://pubmed.ncbi.nlm.nih.gov/25652125/
- Shu D, Li H, Shu Y, \Xiong G, Carson WE, 3rd, et al. Systemic delivery of anti-miRNA for suppression of triple negative breast cancer utilizing RNA nanotechnology. ACS Nano. 2015; 9: 9731–9740. PubMed: https://pubmed.ncbi.nlm.nih.gov/26387848/
- Haussecker D. Current issues of RNAi therapeutics delivery and development. J Control Release. 2014; 195: 49–54. PubMed: https://pubmed.ncbi.nlm.nih.gov/25111131/
- Wang J, Lu Z, Wientjes MG, Au JLS. Delivery of siRNA therapeutics: barriers and carriers. AAPS J. 2010; 12: 492–503. PubMed: https://pubmed.ncbi.nlm.nih.gov/20544328/
- Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, et al.Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. 2005; 11: 263–270. PubMed: https://pubmed.ncbi.nlm.nih.gov/15723075/
- Judge AD, Sood V, Shaw JR, Fang D, McClintock K, et al. Sequencedependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol. 2005; 23: 457–462. PubMed: https://pubmed.ncbi.nlm.nih.gov/15778705/
- Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, et al. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA. 2006; 12: 1179–1187. PubMed: https://pubmed.ncbi.nlm.nih.gov/16682560/
- Hong H, Zhang Y, Cai WB. in vivo imaging of RNA Interference. J Nucl Med. 2010; 51: 169–172. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2832477/
- Willmann JK, van Bruggen N, Dinkelborg LM, Gambhir SS. Molecular imaging in drug development. Nat Rev Drug Discov. 2008; 7: 591–607. PubMed: https://pubmed.ncbi.nlm.nih.gov/18591980/
- Wu SY, Lopez-Berestein G, Calin GA, Sood AK. RNAi therapies: drugging the undruggable. Sci Transl Med. 2014; 6: 240–247. PubMed: https://pubmed.ncbi.nlm.nih.gov/24920658/
- Contag CH, Bachmann MH. Advances in in vivo bioluminescence imaging of gene expression. Annu Rev Biomed Eng. 2002; 4: 235–260. PubMed: https://pubmed.ncbi.nlm.nih.gov/12117758/
- Wang F, Song X, Li X, Xin J, Wang S, et al. Noninvasive visualization of microRNA-16 in the chemoresistance of gastric cancer using a dual reporter gene imaging system. PLoS ONE. 2013; 8: e61792. PubMed: https://pubmed.ncbi.nlm.nih.gov/23613938/
- Huang X, Lee S, Chen X. Design of “smart” probes for optical imaging of apoptosis. Am J Nucl Med Mol Imaging. 2011; 1: 3–17. PubMed: https://pubmed.ncbi.nlm.nih.gov/22514789/
- Sosnovik DE, Weissleder R. Emerging concepts in molecular MRI. Curr Opin Biotechnol. 2007; 18: 4–10. PubMed: https://pubmed.ncbi.nlm.nih.gov/17126545/
- Alauddin MM. Positron emission tomography (PET) imagingwith (18)F-based radiotracers. Am J Nucl Med Mol Imaging. 2012; 2: 55–76. PubMed: https://pubmed.ncbi.nlm.nih.gov/23133802/
- Bhargava P, He G, Samarghandi A, Delpassand ES. Pictorial review of SPECT/CT imaging applications in clinical nuclear medicine. Am J Nucl Med Mol Imaging. 2012; 2: 221–231. PubMed: https://pubmed.ncbi.nlm.nih.gov/23133813/
- Dayton PA, Rychak JJ. Molecular ultrasound imaging using microbubble contrast agents. Front Biosci. 2007; 12: 5124–5142. PubMed: https://pubmed.ncbi.nlm.nih.gov/17569635/
- Pai SI, Lin YY, Macaes B, Meneshian A, Hung CF, et al. Prospects of RNA interference therapy for cancer. Gene Ther. 2006; 13: 464–477. PubMed: https://pubmed.ncbi.nlm.nih.gov/16341059/
- Rao DD, Wang ZH, Senzer N, Nemunaitis J. RNA Interference and personalized cancer therapy. Discov Med. 2013; 81: 101–110. PubMed: https://pubmed.ncbi.nlm.nih.gov/23449112/
- M.D.A.C. Center. EphA2 Gene Targeting Using Neutral Liposomal Small Interfering RNA Delivery (IND# 72924): A Phase I Clinical Trial. https://clinicaltrials.gov/cT2/ show/NCT01591356/term=NCT015913562012
- C.C.C.o.W.F. University, A Phase 1, Open-Label, Dose-Ranging Study to Assess the Safety and Immunologic Activity of APN401. https://clinicaltrials.gov/cT2/show/NCT021662552014
- Pruitt S. Phase I Study of Active Immunotherapy ofMetastaticMelanomawithMature Autologous Dendritic Cells Transfectedwith Tumor Antigen RNA and Small Inhibitory RNAs to Alter Proteasomal Antigen Processing. https://clinicaltrials.gov/cT2/show/NCT006725422008
- S.T. GmbH, A Prospective, Open-label, Single Center, Dose Finding Phase I-Study with Atu027 (an siRNA Formulation) in Subjects with Advanced Solid Cancer. https://clinicaltrials.gov/cT2/show/NCT009385742009
- S. Ltd, Phase I—Escalating Dose Study of siG12D LODER (Local Drug EluteR) in Patients With Locally Advanced Adenocarcinoma of the Pancreas, and a Single Dose Study of siG12D LODER (Local Drug EluteR) in PatientsWith Non-operable Adenocarcinoma of the Pancreas. https://clinicaltrials.gov/cT2/show/NCT011887852010
- N.C.I. (NCI), A Phase 1 Dose Escalation Study of Hepatic Intra-Arterial Administration of TKM 080301 (Lipid Nanoparticles Containing siRNA Against the PLK1 Gene Product) in Patients with Colorectal, Pancreas, Gastric, Breast, Ovarian and Esophageal Cancers with Hepatic. https://clinicaltrials.gov/cT2/show/NCT014370072011
- N.D. Corporation, A Phase 1b/2, Open Label, Randomized, Repeat Dose, Dose Escalation Study to Evaluate the Safety, Tolerability, Biological Activity, and Pharmacokinetics of ND-L02-s0201 Injection, a Vitamin A-coupled Lipid Nanoparticle Containing siRNA Against HSP47, in Subjects with Moderate to Extensive Hepatic Fibrosis (METAVIR F3-4). https://clinicaltrials.gov/cT2/show/NCT022274592014
- Dicerna Pharmaceuticals, Phase I,Multicenter, Cohort Dose Escalation Trial to Determine the Safety, Tolerance, and Maximum Tolerated Dose of DCR-MYC, a Lipid Nanoparticle (LNP)-Formulated Small Inhibitory RNA (siRNA) Oligonucleotide Targeting MYC, in Patients with Refractory Locally Advanced or Metastatic Solid Tumor Malignancies, Multiple Myeloma, or Lymphoma. https://clinicaltrials.gov/cT2/show/NCT021105632014
- C. Pharmaceuticals, A Phase I, Dose-Escalating Study of the Safety of Intravenous CALAA-01 in Adults with Solid Tumors Refractory to Standard-of-Care Therapies. https://clinicaltrials.gov/cT2/show/NCT006890652008
- T.P. Corporation, A Phase 1/2 Dose Escalation Study to Determine the Safety, Pharmacokinetics, and Pharmacodynamics of Intravenous TKM-080301 in Patients with Advanced Solid Tumors. https://clinicaltrials.gov/cT2/show/NCT012622352010
- Pharmaceuticals, A Multi-Center, Open Label, Phase 1 Dose-Escalation Trial to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Intravenous ALN-VSP02 in Patients with Advanced Solid Tumors with Liver Involvement. https://clinicaltrials.gov/cT2/show/NCT008821802009
- Pharmaceuticals, A Multi-center, Open-Label, Extension Study of ALN-VSP02 in Cancer Patients Who Have Responded to ALN-VSP02 Treatment. https://clinicaltrials.gov/cT2/show/NCT011580792009
- Aleku SM, Schulz P, Keil O, Santel A, Schaeper U, et al. Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression. Cancer Res. 2008; 68: 9788–9798. PubMed: https://pubmed.ncbi.nlm.nih.gov/19047158/
- Aleku SM, Roder N, Mopert K, Durieux B, Janke O, et al. Atu027 prevents pulmonary metastasis in experimental and spontaneous mouse metastasis models. Clin Cancer Res. 2010; 16: 5469–5480. PubMed: https://pubmed.ncbi.nlm.nih.gov/21062934/
- Leenders F, Mopert K, Schmiedeknecht A, Santel A, Czauderna F, et al. PKN3 is required formalignant prostate cell growth downstream of activated PI 3-kinase. EMBO J. 2004; 23: 3303–3313. PubMed: https://pubmed.ncbi.nlm.nih.gov/15282551/
- Strumberg D, Schultheis B, Traugott U, Vank C, Santel A, et al. Phase I clinical development of Atu027, a siRNA formulation targeting PKN3 in patients with advanced solid tumors. Int J Clin Pharmacol Ther. 2012; 50: 76–78. PubMed: https://pubmed.ncbi.nlm.nih.gov/22192654/
- Schultheis B, Strumberg D, Santel A, Vank C, Gebhardt F, et al. First-in-human phase I study of the liposomal RNA Interference therapeutic Atu027 in patients with advanced solid tumors. J Clin Oncol. 2014; 32: 4141–4148. PubMed: https://pubmed.ncbi.nlm.nih.gov/25403217/
- Blangy A, Lane HA, d'Herin P, Harper M, Kress M, et al. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995; 83: 1159–1169. PubMed: https://pubmed.ncbi.nlm.nih.gov/8548803/
- Itakura J, Ishiwata T, Shen B, Kornmann M, Korc M. Concomitant overexpression of vascular endothelial growth factor and its receptors in pancreatic cancer. Int J Cancer. 2000; 85: 27–34. PubMed: https://pubmed.ncbi.nlm.nih.gov/10585578/
- Boocock CA, Charnockjones DS, Sharkey AM, Mclaren J, Barker PJ, et al. Expression of vascular endothelial growth-factor and its receptors Flt and Kdr in ovarian-carcinoma. J Natl Cancer Inst. 1995; 87: 506–516. PubMed: https://pubmed.ncbi.nlm.nih.gov/7707437/
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003; 9: 669–676. PubMed: https://pubmed.ncbi.nlm.nih.gov/12778165/
- Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. RNA interference targeting the M2 subunit of ribonucleotide reductase enhances pancreatic adenocarcinoma chemosensitivity to gemcitabine, Oncogene. 2004; 23: 1539–1548. PubMed: https://pubmed.ncbi.nlm.nih.gov/14661056/
- Wang YX, Ng CK. The impact of quantitative imaging in medicine and surgery: charting our course for the future. Quant Imaging Med Surg. 2011; 1: 1–3. PubMed: https://pubmed.ncbi.nlm.nih.gov/23256048/
- Tracy Zimmermann VK, Harrop J, Chan A, Chiesa J, Peters G, et al. Phase I First-in-Human Trial of ALN-TTRsc, a Novel RNA Interference Therapeutic for the Treatment of Familial Amyloidotic Cardiomyopathy (FAC). http://www.alnylam.com/web/wp-content/uploads/2013/09/ALN-TTRsc-PhI-HFSA-Poster-Sep2013.pdf2013
- Buyens K, De Smedt SC, Braeckmans K, Demeester J, Peeters L, et al. Liposome based systems for systemic siRNA delivery: stability in blood sets the requirements for optimal carrier design. J Control Release. 2012; 158: 362–370. PubMed: https://pubmed.ncbi.nlm.nih.gov/22023849/
- Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013; 12: 967–977.
- Oh YK, Park TG. siRNA delivery systems for cancer treatment. Adv Drug Deliv Rev. 2009; 61: 850–862. PubMed: https://pubmed.ncbi.nlm.nih.gov/19422869/
- Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, et al. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014; 15: 541–555. PubMed: https://pubmed.ncbi.nlm.nih.gov/25022906/
- Zhang P, Chen Y, Zeng Y, Shen C, Li R, et al. Virus-mimetic nanovesicles as a versatile antigen-delivery system. Proc Natl Acad Sci. U S A. 2015; 112: E6129–E6138. PubMed: https://pubmed.ncbi.nlm.nih.gov/26504197/
- Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what Is the appropriate Target? Theranostics. 2014; 4: 81–89. PubMed: https://pubmed.ncbi.nlm.nih.gov/24396516/
- Matsumura Y, Maeda H, A new concept for macromolecular therapeutics in cancer- chemotherapy—mechanism of tumoritropic accumulation of proteins and the antitumor agent Smancs. Cancer Res. 1996; 46: 6387–6392. PubMed: https://pubmed.ncbi.nlm.nih.gov/2946403/
- Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001; 53: 283–318. PubMed: https://pubmed.ncbi.nlm.nih.gov/11356986/
- Cho KJ, Wang X, Nie SM, Chen Z, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008; 14: 1310–1316. PubMed: https://pubmed.ncbi.nlm.nih.gov/18316549/
- van Vlerken LE, Vyas TK, Amiji MM. Poly(ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery. Pharm Res. 2007; 24: 1405–1414. PubMed: https://pubmed.ncbi.nlm.nih.gov/17393074/
- Maclachlan I, Cullis P. Diffusible-PEG-lipid stabilized plasmid lipid particles. Adv Genet. 2005; 157: 188 53pa. PubMed: https://pubmed.ncbi.nlm.nih.gov/16243063/
- Vaupel P, Kallinowski F, Okunieff P. Blood-flow, oxygen and nutrient supply, and metabolic microenvironment of human-tumors—a review. Cancer Res. 1989; 49: 6449–6465. PubMed: https://pubmed.ncbi.nlm.nih.gov/2684393/
- Li L, Wang R, Wilcox D, Zhao X, Song J, et al. Tumor vasculature is a key determinant for the efficiency of nanoparticlemediated siRNA delivery. Gene Ther. 2012; 19: 775–780. PubMed: https://pubmed.ncbi.nlm.nih.gov/21956688/
- Huang K, Ma H, Liu J, Huo S, Kumar A, et al. Size-dependent localization and penetration of ultrasmall gold nanoparticles in cancer cells, multicellular spheroids, and tumors in vivo. ACS Nano. 2012; 6: 4483–4493. PubMed: https://pubmed.ncbi.nlm.nih.gov/22540892/
- Cunha L, Szigeti K, Mathe D, Metello LF. The role ofmolecular imaging inmodern drug development. Drug Discov Today. 2014; 19: 936–948. PubMed: https://pubmed.ncbi.nlm.nih.gov/24434047/
- Meade BR, Dowdy SF. Exogenous siRNA delivery using peptide transduction domains/ cell penetrating peptides. Adv Drug Deliv Rev. 2007; 59: 134–140. PubMed: https://pubmed.ncbi.nlm.nih.gov/17451840/
- Rozema DB, Lewis DL, Wakefield DH, Wong SC, Klein JJ, et al. Dynamic polyconjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc Natl Acad Sci. U S A. 2007; 104: 12982–12987. PubMed: https://pubmed.ncbi.nlm.nih.gov/17652171/
- Cardoso AL, Simoes S, de Almeida LP, Pelisek J, Culmsee C, et al. siRNA delivery by a transferrin-associated lipid-based vector: a non-viral strategy to mediate gene silencing. J Gene Med. 2007; 9: 170–183. PubMed: https://pubmed.ncbi.nlm.nih.gov/17351968/
- Chu TC, Twu KY, Ellington AD, Levy M. Aptamer mediated siRNA delivery. Nucleic Acids Res. 2006; 34: e73. PubMed: https://pubmed.ncbi.nlm.nih.gov/16740739/
- Lorenzer C, Dirin M, Winkler AM, Baumann V, Winkler J. Going beyond the liver: progress and challenges of targeted delivery of siRNA therapeutics. J Control Release. 2015; 203: 1–15. PubMed: https://pubmed.ncbi.nlm.nih.gov/25660205/
- Gruenberg J, van der Goot FG. Mechanisms of pathogen entry through the endosomal compartments. Nat Rev Mol Cell Biol. 2006; 7: 495–504. PubMed: https://pubmed.ncbi.nlm.nih.gov/16773132/
- Sonawane ND, Szoka FC, Jr., Verkman AS. Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine-DNA polyplexes. J Biol Chem. 2003; 278: 44826–44831. PubMed: https://pubmed.ncbi.nlm.nih.gov/12944394/
- Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci. U S A. 1995; 92: 7297–7301. PubMed: https://pubmed.ncbi.nlm.nih.gov/7638184/
- Qiu S, Adema CM, Lane T. A computational study of off-target effects of RNA interference. Nucleic Acids Res. 2005; 33: 1834–1847. PubMed: https://pubmed.ncbi.nlm.nih.gov/15800213/
- Goodchild A, Nopper N, King A, Doan T, Tanudji M, et al. Sequence determinants of innate immune activation by short interfering RNAs. BMC Immunol. 2009; 10: 40. PubMed: https://pubmed.ncbi.nlm.nih.gov/19630977/
- Judge AD, Sood V, Shaw JR, Fang D, McClintock K, et al. Sequence dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol. 2005; 23: 457–462. PubMed: https://pubmed.ncbi.nlm.nih.gov/15778705/
- Sioud M. Does the understanding of immune activation by RNA predict the design of safe siRNAs? Front Biosci. 2008; 13: 4379–4392. PubMed: https://pubmed.ncbi.nlm.nih.gov/18508517/
- Sevick-Muraca EM, Houston JP, Gurfinkel M. Fluorescence-enhanced, near infrared diagnostic imaging with contrast agents. Curr Opin Chem Biol. 2002; 6: 642–650. PubMed: https://pubmed.ncbi.nlm.nih.gov/12413549/
- Frangioni JV. in vivo near-infrared fluorescence imaging. Curr Opin Chem Biol. 2003; 7: 626–634. PubMed: https://pubmed.ncbi.nlm.nih.gov/14580568/
- Zanzonico P. Noninvasive imaging for supporting basic research, Small Animal Imaging: Basics and Practical Guide. 2011; 3–16.
- Leblond F, Davis SC, Valdés PA, Pogue BW. Pre-clinical whole-body fluorescence imaging: review of instruments, methods and applications. J Photochem Photobiol. 2010; 98: 77–94. PubMed: https://pubmed.ncbi.nlm.nih.gov/20031443/
- Guo Z, Park S, Yoon J, Shin I. Recent progress in the development of near-infrared fluorescent probes for bioimaging applications. Chem Soc Rev. 2014; 43: 16–29.
- Kim SH, Jeong JH, Lee SH, Kim SW, Park TG. Local and systemic delivery of VEGF siRNA using polyelectrolyte complex micelles for effective treatment of cancer. J Control Release. 2008; 129: 107–116. PubMed: https://pubmed.ncbi.nlm.nih.gov/18486981/
- Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013; 31: 638–646. PubMed: https://pubmed.ncbi.nlm.nih.gov/23792630/
- Chang E, Zhu MQ, Drezek R. Novel siRNA-based molecular beacons for dual imaging and therapy. Biotechnol J. 2007; 2: 422–425. PubMed: https://pubmed.ncbi.nlm.nih.gov/17330221/
- Christie RJ, Matsumoto Y, Miyata K, Nomoto T, Fukushima S, et al. Targeted polymeric micelles for siRNA treatment of experimental cancer by intravenous injection. ACS Nano. 2012; 6: 5174–5189. PubMed: https://pubmed.ncbi.nlm.nih.gov/22575090/
- Christie RJ, Miyata K, Matsumoto Y, Nomoto T, Menasco D, et al. Effect of polymer structure on micelles formed between siRNA and cationic block copolymer comprising thiols and amidines. Biomacromolecules. 2011; 12: 3174–3185. PubMed: https://pubmed.ncbi.nlm.nih.gov/21863796/
- Oe Y, Christie RJ, Naito M, Low SA, Fukushima S, et al. Actively-targeted polyion complex micelles stabilized by cholesterol and disulfide cross-linking for systemic delivery of siRNA to solid tumors. Biomaterials. 2014; 35: 7887–7895. PubMed: https://pubmed.ncbi.nlm.nih.gov/24930854/
- Chen AA, Derfus AM, Khetani SR, Bhatia SN. Quantum dots tomonitor RNAi delivery and improve gene silencing. Nucleic Acids Res. 2005; 33: e190. PubMed: https://pubmed.ncbi.nlm.nih.gov/16352864/
- Wang J, Zhang P, Huang C, Liu G, Leung KC, et al. High performance photoluminescent carbon dots for in vitro and in vivo bioimaging: effect of nitrogen doping ratios. Langmuir. 2015; 31: 8063–8073. PubMed: https://pubmed.ncbi.nlm.nih.gov/26135003/
- Jiang S, Zhang Y, Lim KM, Sim EKW, Ye L. NIR-to-visible upconversion nanoparticles for fluorescent labeling and targeted delivery of siRNA. Nanotechnology. 2009; 20: e155101. PubMed: https://pubmed.ncbi.nlm.nih.gov/19420539/
- Jung J, Solanki A, Memoli KA, Kamei K, Kim H, et al. Selective inhibition of human brain tumor cells through multifunctional quantum-dot-based siRNA delivery. Angew Chem Int Ed. 2010; 49: 103–107. PubMed: https://pubmed.ncbi.nlm.nih.gov/19950159/
- Wang J, Liu G, Leung KC, Loffroy R, Lu PX, et al. Opportunities and challenges of fluorescent carbon dots in translational optical imaging. Curr Pharm Des. 2015; 21: 5401–5416. PubMed: https://pubmed.ncbi.nlm.nih.gov/26377656/
- Wang L, Wang X, Bhirde A, Cao J, Zeng Y, et al. Carbon- dot-based two-photon visible nanocarriers for safe and highly efficient delivery of siRNA and DNA. Adv Health Mater. 2014; 3: 1203–1209. PubMed: https://pubmed.ncbi.nlm.nih.gov/24692418/
- Chatterjee DK, Gnanasammandhan Y, Fau-Zhang MK, Zhang Y. Small upconverting fluorescent nanoparticles for biomedical applications. Small. 2010; 6: 2781–2795. PubMed: https://pubmed.ncbi.nlm.nih.gov/21064086/
- Wang L, Liu J, Dai Y, Yang Q, Zhang Y, et al. Efficient gene delivery and multimodal imaging by lanthanide-based upconversion nanoparticles. Langmuir. 2014; 30: 13042–13051. PubMed: https://pubmed.ncbi.nlm.nih.gov/25291048/
- Nayak TR, Krasteva LK, Cai W. Multimodality imaging of RNA Interference. Curr Med Chem. 2013; 20: 3664–3675. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3749287/
- McCaffrey AP, Meuse L, Pham TT, Conklin DS, Hannon GJ, et al. RNA interference in adult mice. Nature. 2002; 418: 38–39. PubMed: https://pubmed.ncbi.nlm.nih.gov/12097900/
- Lewis DL, Hagstrom JE, Loomis AG, Wolff JA, Herweijer H. Efficient delivery of siRNA for inhibition of gene expression in postnatal mice. Nat Genet. 2002; 32: 107–108. PubMed: https://pubmed.ncbi.nlm.nih.gov/12145662/
- Pichler A, Zelcer N, Prior JL, Kuil AJ, Piwnica-Worms D. In vivo RNA interference-mediated ablation of MDR1 P-glycoprotein. Clin Cancer Res. 2005; 11: 4487–4494. PubMed: https://pubmed.ncbi.nlm.nih.gov/15958634/
- Pittella F, Cabral H, Maeda Y, Mi P, Watanabe S, et al. Systemic siRNA delivery to a spontaneous pancreatic tumor model in transgenic mice by PEGylated calcium phosphate hybrid micelles. J Control Release. 2014; 178: 18–24. PubMed: https://pubmed.ncbi.nlm.nih.gov/24440662/
- Bartlett DW, Davis ME. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Res. 2006; 34: 322–333. PubMed: https://pubmed.ncbi.nlm.nih.gov/16410612/
- Kim SH, Mok H, Jeong JH, Kim SW, Park TG. Comparative evaluation of targetspecific GFP gene silencing efficiencies for antisense ODN, synthetic siRNA, and siRNA plasmid complexed with PEI-PEG-FOL conjugate. Bioconjug Chem. 2006; 17: 241–244. PubMed: https://pubmed.ncbi.nlm.nih.gov/16417275/
- Huschka R, Barhoumi A, Liu Q, Roth JA, Ji L, et al. Gene silencing by gold nanoshell-mediated delivery and laser-triggered release of antisense oligonucleotide and siRNA. ACS Nano. 2012; 6: 7681–7691. PubMed: https://pubmed.ncbi.nlm.nih.gov/22862291/
- James ML, Gambhir SS. A molecular imaging primer: modalities, imaging agents, and applications. Physiol Rev. 2012; 92: 897–965. PubMed: https://pubmed.ncbi.nlm.nih.gov/22535898/
- Phelps ME. Positron emission tomography provides molecular imaging of biological processes. Proc Natl Acad Sci U S A. 2000; 97: 9226–9233. PubMed: https://pubmed.ncbi.nlm.nih.gov/10922074/
- Ziegler SI. Positron emission tomography: principles, technology, and recent developments. Nucl Phys A. 2005; 752: 679c–687c.
- Lammers T, Aime S, Hennink WE, Storm G, Kiessling F. Theranostic nanomedicine. Acc Chem Res. 2011; 44: 1029–1038. PubMed: https://pubmed.ncbi.nlm.nih.gov/21545096/
- Lu W, Zhang G, Zhang R, Flores LG,2nd, Huang Q, et al. Tumor sitespecific silencing of NF-kappaB p65 by targeted hollow gold nanosphere-mediated photothermal transfection. Cancer Res. 2010; 70: 3177–3188. PubMed: https://pubmed.ncbi.nlm.nih.gov/20388791/
- Nayak TR, Krasteva LK, Cai W. Multimodality imaging of RNA interference. Curr Med Chem. 2013; 20: 3664–3675. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3749287/
- Hatanaka K, Asai T, Koide H, Kenjo E, Tsuzuku T, et al. Development of double-stranded siRNA labeling method using positron emitter and its in vivo trafficking analyzed by positron emission tomography. Bioconjug Chem. 2010; 21: 756–763.
- Lu FM, Yuan Z. PET/SPECT molecular imaging in clinical neuroscience: recent advances in the investigation of CNS diseases. Quant Imaging Med Surg. 2015; 5: 433–447. PubMed: https://pubmed.ncbi.nlm.nih.gov/26029646/
- Liu N, Ding H, Vanderheyden JL, Zhu Z, Zhang Y. Radiolabeling small RNA with technetium-99 m for visualizing cellular delivery and mouse biodistribution. Nucl Med Biol. 2007; 34: 399–404. PubMed: https://pubmed.ncbi.nlm.nih.gov/17499729/
- Merkel OM, Librizzi D, Pfestroff A, Schurrat T, Behe M, et al. In vivo SPECT and real-time gamma camera imaging of biodistribution and pharmacokinetics of siRNA delivery using an optimized radiolabeling and purification procedure. Bioconjug Chem. 2009; 20: 174–182. PubMed: https://pubmed.ncbi.nlm.nih.gov/19093855/
- Faulkner W. Basic Principles of MRIOutSource 1996.
- Wang YX. Super paramagnetic iron oxide based MRI contrast agents: current status of clinical application. Quant Imaging Med Surg. 2011; 1: 35–40. PubMed: https://pubmed.ncbi.nlm.nih.gov/23256052/
- Mok H, Veiseh O, Fang C, Kievit FM, Wang FY, et al. pH-sensitive siRNA nanovector for targeted gene silencing and cytotoxic effect in cancer cells. Mol Pharm. 2010; 7: 1930–1939. PubMed: https://pubmed.ncbi.nlm.nih.gov/20722417/
- Shen M, Gong F, Pang P, Zhu K, Meng X, et al. An MRI-visible non-viral vector for targeted Bcl-2 siRNA delivery to neuroblastoma. Int J Nanomed. 2012; 7: 3319–3332. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3396391/
- Li C, Penet MF, Wildes F, Takagi T, Chen ZH, et al. Nanoplex delivery of siRNA and prodrug enzyme for multimodality image-guided molecular pathway targeted cancer therapy. ACS Nano. 2010; 4: 6707–6716. PubMed: https://pubmed.ncbi.nlm.nih.gov/20958072/
- Bae KH, Lee K, Kim C, Park TG. Surface functionalized hollow manganese oxide nanoparticles for cancer targeted siRNA delivery and magnetic resonance imaging. Biomaterials. 2011; 32: 176–184. PubMed: https://pubmed.ncbi.nlm.nih.gov/20934746/
- Zhou Z, Huang D, Bao J, Chen Q, Liu G, et al. A synergistically enhanced T(1)–T(2) dual-modal contrast agent. Adv Mater. 2012; 24: 6223–6228. PubMed: https://pubmed.ncbi.nlm.nih.gov/22972529/
- Wang X, Zhou Z, Wang Z, Xue Y, Zeng Y, et al. Gadolinium embedded iron oxide nanoclusters as T1–T2 dual-modal MRI-visible vectors for safe and efficient siRNA delivery. Nanoscale. 2013; 5: 8098–8104. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3775844/
- Liang HD, Blomley MJK. The role of ultrasound inmolecular imaging. Br J Radiol. 2003; 76: S140–S150. https://www.ncbi.nlm.nih.gov/pubmed/15572336/
- Shim MS, Kwon YJ. Stimuli-responsive polymers and nanomaterials for gene delivery and imaging applications. Adv Drug Deliv Rev. 2012; 64: 1046–1059. PubMed: https://pubmed.ncbi.nlm.nih.gov/22329941/
- Unger, Porter T, Lindner J, Grayburn P. Cardiovascular drug delivery with ultrasound and microbubbles. Adv Drug Deliv Rev. 2014; 72: 110–126. PubMed: https://pubmed.ncbi.nlm.nih.gov/24524934/
- Martin KH, Dayton PA. Current status and prospects for microbubbles in ultrasound theranostics, Wiley Interdiscip. Rev Nanomed Nanobiotechnol. 2013; 5: 329–345. PubMed: https://pubmed.ncbi.nlm.nih.gov/23504911/
- Florinas S, Kim J, Nam K, Janat-Amsbury MM, Kim SW. Ultrasound-assisted siRNA delivery via arginine-grafted bioreducible polymer and microbubbles targeting VEGF for ovarian cancer treatment. J Control Release. 183: 1–8. PubMed: https://pubmed.ncbi.nlm.nih.gov/24657947/
- Vandenbroucke RE, Lentacker I, Demeester J, De Smedt SC, et al. Ultrasound assisted siRNA delivery using PEG-siPlex loaded microbubbles. J Control Release. 2008; 126: 265–273. PubMed: https://pubmed.ncbi.nlm.nih.gov/18237813/
- Liu Y, Miyoshi H, Nakamura M. Encapsulated ultrasoundmicrobubbles: therapeutic application in drug/gene delivery. J Control Release. 2006; 114: 89–99. PubMed: https://pubmed.ncbi.nlm.nih.gov/16824637/
- Chen CC, Sheeran PS, Wu SY, Olumolade OO, Dayton PA, et al. Targeted drug delivery with focused ultrasound-induced blood–brain barrier opening using acoustically-activated nanodroplets. J Control Release. 2013; 172: 795–804. PubMed: https://pubmed.ncbi.nlm.nih.gov/24096019/
- Cheng X, Li H, Chen Y, Luo B, Liu X, et al. Ultrasound-triggered phase transition sensitive magnetic fluorescent nanodroplets as a multimodal imaging contrast agent in rat and mouse model. PLoS ONE. 2013; 8: e85003. PubMed: https://pubmed.ncbi.nlm.nih.gov/24391983/
- Dayton PA, Zhao S, Bloch SH, Schumann P, Penrose K, et al. Application of ultrasound to selectively localize nanodroplets for targeted imaging and therapy. Mol Imaging. 2006; 5: 160–174. PubMed: https://pubmed.ncbi.nlm.nih.gov/16954031/
- Jian J, Liu C, Gong Y, Su L, Zhang B, et al. India ink incorporated multifunctional phase-transition nanodroplets for photoacoustic/ultrasound dual-modality imaging and photoacoustic effect based tumor therapy. Theranostics. 2014; 4: 1026–1038. PubMed: https://pubmed.ncbi.nlm.nih.gov/25161702/
- Horie S, Watanabe Y, Ono M, Mori S, Kodama T. Evaluation of antitumor effects following tumor necrosis factor-alpha gene delivery using nanobubbles and ultrasound. Cancer Sci. 2011; 102: 2082–2089. PubMed: https://pubmed.ncbi.nlm.nih.gov/21824220/
- Wang CH, Huang YF, Yeh CK. Aptamer-conjugated nanobubbles for targeted ultrasound molecular imaging. Langmuir. 2011; 27: 6971–6976. PubMed: https://pubmed.ncbi.nlm.nih.gov/21553884/
- Yin T, Wang P, Li J, Wang Y, Zheng B, et al. Tumorpenetrating codelivery of siRNA and paclitaxel with ultrasound-responsive nanobubbles hetero-assembled from polymeric micelles and liposomes. Biomaterials. 2014; 35: 5932–5943. PubMed: https://pubmed.ncbi.nlm.nih.gov/24746965/
- Chen Z, Liang K, Liu J, Xie M, Wang X, et al. Enhancement of survivin gene downregulation and cell apoptosis by a novel combination: liposome microbubbles and ultrasound exposure. Med Oncol. 2009; 26: 491–500. PubMed: https://pubmed.ncbi.nlm.nih.gov/19137432/
- Castle J, Butts M, Healey A, Kent K, Marino M, et al.Ultrasoundmediated targeted drug delivery: recent success and remaining challenges. Am J Physiol Heart Circ Physiol. 2013; 304: H350–H357. PubMed: https://pubmed.ncbi.nlm.nih.gov/23203969/
- Kaneko OF, Willmann JK. Ultrasound for molecular imaging and therapy in cancer. Quant Imaging Med Surg. 2012; 2: 87–97. PubMed: https://pubmed.ncbi.nlm.nih.gov/23061039/
- Son S, Min HS, You DG, Kim BS, Kwon IC. Echogenic nanoparticles for ultrasound technologies: evolution from diagnostic imaging modality to multimodal theranostic agent. Nano Today. 2012; 9: 525–540. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4672021/ V
- Sboros V. Response of contrast agents to ultrasound. Adv Drug Deliv Rev. 2008; 60: 1117–1136. PubMed: https://pubmed.ncbi.nlm.nih.gov/18486270/
- Carson AR, McTiernan CF, Lavery L, Grata M, Leng X, et al. Ultrasound-targeted microbubble destruction to deliver siRNA cancer therapy. Cancer Res. 2012; 72: 6191–6199. PubMed: https://pubmed.ncbi.nlm.nih.gov/23010078/
- Zhou QL, Chen ZY, Wang YX, Yang F, Lin Y, et al. Ultrasound-mediated local drug and gene delivery using nanocarriers. Biomed Res Int. 2014; e963891. PubMed: https://pubmed.ncbi.nlm.nih.gov/25202710/
- Yin T, Wang P, Li J, Zheng R, Zheng B, et al. Ultrasoundsensitive siRNA-loaded nanobubbles formed by hetero-assembly of polymeric micelles and liposomes and their therapeutic effect in gliomas. Biomaterials. 2013; 34: 4532–4543. PubMed: https://pubmed.ncbi.nlm.nih.gov/23522375/
- Lee DE, Koo H, Sun IC, Ryu TH, Kim K, et al. Multifunctional nanoparticles for multimodal imaging and theragnosis. Chem Soc Rev. 2012; 41: 2656–2672. PubMed: https://pubmed.ncbi.nlm.nih.gov/22189429/
- Choi HS, Frangioni JV. Nanoparticles for biomedical imaging: fundamentals of clinical translation. Mol Imaging. 2010; 9: 291–310. PubMed: https://pubmed.ncbi.nlm.nih.gov/21084027/
- Louie AY. Multimodality imaging probes: design and challenges. Chem Rev. 2010; 110: 3146–3195. PubMed: https://pubmed.ncbi.nlm.nih.gov/20225900/
- Boerman OC, Oyen WJG. Multimodality probes: amphibian cars for molecular imaging. J Nucl Med. 2008; 49: 1213–1214. PubMed: https://pubmed.ncbi.nlm.nih.gov/18632805/
- Idee JM, Louguet S, Ballet S, Corot C0 Theranostics and contrast-agents for medical imaging: a pharmaceutical company viewpoint. Quant Imaging Med Surg. 2013; 3: 292–297. PubMed: https://pubmed.ncbi.nlm.nih.gov/24404442/
- Wang YX. Physical scientists research biomedicine: a call for caution. Chin J Cancer Res. 2015; 27: 94–95. PubMed: https://pubmed.ncbi.nlm.nih.gov/25717232/
- Medarova Z, Pham W, Farrar C, Petkova V, Moore A. in vivo imaging of siRNA delivery and silencing in tumors. Nat Med. 2007; 13: 372–377. PubMed: https://pubmed.ncbi.nlm.nih.gov/17322898/
- Mikhaylova M, Stasinopoulos I, Kato Y, Artemov D, Bhujwalla ZM, Imaging of cationic multifunctional liposome-mediated delivery of COX-2 siRNA. Cancer Gene Ther. 2009; 16: 217–226. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5226392/
- Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Impact of tumorspecific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci U S A. 2007; 104: 15549–15554. PubMed: https://pubmed.ncbi.nlm.nih.gov/17875985/
- Lammers T, Aime S, Hennink WE, Storm G, Kiessling F. Theranostic nanomedicine. Acc Chem Res. 2011; 44: 1029–1038. PubMed: https://pubmed.ncbi.nlm.nih.gov/21545096/
- Ma X, Zhao Y, Liang XJ, Theranostic nanoparticles engineered for clinic and pharmaceutics. Acc Chem Res. 2011; 44: 1114–1122. PubMed: https://pubmed.ncbi.nlm.nih.gov/21732606/
- Ryu JH, Koo H, Sun IC, Yuk SH, Choi K, et al. Tumor targetingmultifunctional nanoparticles for theragnosis: new paradigm for cancer therapy. Adv Drug Deliv Rev. 2012; 64: 1447–1458. PubMed: https://pubmed.ncbi.nlm.nih.gov/22772034/
- Lee DE, Koo H, Sun IC, Ryu JH, Kim K, et al. Multifunctional nanoparticles for multimodal imaging and theragnosis. Chem Soc Rev. 2012; 41: 2656–2672. PubMed: https://pubmed.ncbi.nlm.nih.gov/22189429/
- Lee N, Choi SH, Hyeon T. Nano-sized CT contrast agents. Adv Mater. 2013; 25: 2641–2660.
- Hallouard F, Anton N, Choquet P, Constantinesco A, Vandamme T. Iodinated blood pool contrast media for preclinical X-ray imaging applications–a review. Biomaterials. 2010; 31: 6249–6268. PubMed: https://pubmed.ncbi.nlm.nih.gov/20510444/
- Liu Y, Ai K, Lu L. Nanoparticulate X-ray computed tomography contrast agents: from design validation to in vivo applications. Acc Chem Res. 2012; 45: 1817–1827. https://pubs.acs.org/doi/abs/10.1021/ar300150c
- Lusic H, Grinstaff MW. X-ray-computed tomography contrast agents. Chem Rev. 2012; 113: 1641–1666. PubMed: https://pubmed.ncbi.nlm.nih.gov/23210836/
- Jin E, Lu ZR. Biodegradable iodinated polydisulfides as contrast agents for CT angiography. Biomaterials. 2014; 35: 5822–5829. PubMed: https://pubmed.ncbi.nlm.nih.gov/24768156/
- Choi KY, Liu G, Lee S, Chen X. Theranostic nanoplatforms for simultaneous cancer imaging and therapy: current approaches and future perspectives. Nanoscale. 2012; 4: 330–342. PubMed: https://pubmed.ncbi.nlm.nih.gov/22134683/
- Anton N, Vandamme TF. Nanotechnology for computed tomography: a real potential recently disclosed. Pharm Res. 2012; 31: 20–34. PubMed: https://pubmed.ncbi.nlm.nih.gov/23896738/
- Cormode DP, Naha PC, Fayad ZA. Nanoparticle contrast agents for computed tomography: a focus on micelles. Contrast Media Mol Imaging. 2014; 9: 37–52. PubMed: https://pubmed.ncbi.nlm.nih.gov/24470293/
- Li X, Anton N, Zuber G, Vandamme T. Contrast agents for preclinical targeted X-ray imaging. Adv Drug Deliv Rev. 2014; 76: 116–133. PubMed: https://pubmed.ncbi.nlm.nih.gov/25086373/
- Lee JY, Chung SJ, Cho HJ, Kim DD. Iodinated hyaluronic acid oligomer-based nanoassemblies for tumor-targeted drug delivery and cancer imaging. Biomaterials. 2016; 85: 218–231. PubMed: https://pubmed.ncbi.nlm.nih.gov/26874284/
- Deng H, Zhong Y, Du M, Liu Q, Fan Z, et al. Theranostic self-assembly structure of gold nanoparticles for NIR photothermal therapy and X-ray computed tomography imaging. Theranostics. 2014; 4: 904–918. PubMed: https://pubmed.ncbi.nlm.nih.gov/25057315/
- Zhu J, Zheng L, Wen S, Tang Y, Shen M, et al. Targeted cancer theranostics using alpha-tocopheryl succinate-conjugated multifunctional dendrimer-entrapped gold nanoparticles. Biomaterials. 2014; 35: 7635–7646. PubMed: https://pubmed.ncbi.nlm.nih.gov/24927683/
- Jiang Y, Chen J, Deng C, Suuronen EJ, Zhong Z. Click hydrogels, microgels and nanogels: emerging platforms for drug delivery and tissue engineering. Biomaterials. 2011; 35: 4969–4985. PubMed: https://pubmed.ncbi.nlm.nih.gov/24674460/
- Kabanov AV, Vinogradov SV. Nanogels as pharmaceutical carriers: finite networks of infinite capabilities. Angew Chem Int Ed. 2009; 48: 5418–5429. PubMed: https://pubmed.ncbi.nlm.nih.gov/19562807/
- Zhang X, Malhotra S, Molina M, Haag R. Micro-and nanogels with labile crosslinksfrom synthesis to biomedical applications. Chem Soc Rev. 2015; 44: 1948–1973.
- Chen W, Achazi K, Schade B, Haag R. Charge-conversional and reduction-sensitive poly (vinyl alcohol) nanogels for enhanced cell uptake and efficient intracellular doxorubicin release. J Control Release. 2015; 205: 15–24. PubMed: https://pubmed.ncbi.nlm.nih.gov/25445693/
- Morimoto N, Hirano S, Takahashi H, Loethen S, Thompson DH, et al. Selfassembled pH-sensitive cholesteryl pullulan nanogel as a protein delivery vehicle. Biomacromolecules. 2013; 14: 56–63. PubMed: https://pubmed.ncbi.nlm.nih.gov/23215439/
- Jiang L, Zhou Q, Mu K, Xie H, Zhu Y, et al. pH/temperature sensitive magnetic nanogels conjugated with Cy5.5-labled lactoferrin for MR and fluorescence imaging of glioma in rats. Biomaterials. 2013; 34: 7418–7428. PubMed: https://pubmed.ncbi.nlm.nih.gov/23810255/
- Steinhilber D, Witting M, Zhang X, Staegemann M, Paulus F, et al. Surfactant free preparation of biodegradable dendritic polyglycerol nanogels by inverse nanoprecipitation for encapsulation and release of pharmaceutical biomacromolecules. J Control Release. 2013; 169: 289–295. PubMed: https://pubmed.ncbi.nlm.nih.gov/23262202/
- Yang H, Wang Q, Chen W, Zhao Y, Yong T, et al. Hydrophilicity/ hydrophobicity reversable and redox-sensitive nanogels for anticancer drug delivery. Mol Pharm. 2015; 12: 1636–1647. PubMed: https://pubmed.ncbi.nlm.nih.gov/25833093/
- Zhang X, Achazi K, Steinhilber D, Kratz F, Dernedde J, et al. A facile approach for dual-responsive prodrug nanogels based on dendritic polyglycerols with minimal leaching. J Control Release. 2014; 174: 209–216. PubMed: https://pubmed.ncbi.nlm.nih.gov/24225227/
- Yang C, Wang X, Yao X, Zhang Y, Wu W, et al. Hyaluronic acid nanogels with enzyme-sensitive cross-linking group for drug delivery. J Control Release. 2015; 205: 206–217. PubMed: https://pubmed.ncbi.nlm.nih.gov/25665867/
- Chen W, Zheng M, Meng F, Cheng R, Deng C, et al. In situ forming reduction-sensitive degradable nanogels for facile loading and triggered intracellular release of proteins. Biomacromolecules. 3; 14: 1214–1222. PubMed: https://pubmed.ncbi.nlm.nih.gov/23477570/
- Wei X, Senanayake TH, Warren G, Vinogradov SV. Hyaluronic acid-based nanogel-drug conjugates with enhanced anticancer activity designed for the targeting of CD44-positive and drug-resistant tumors. Bioconjug Chem. 2013; 24: 658–668. PubMed: https://pubmed.ncbi.nlm.nih.gov/23547842/
- Stefanello TF, Szarpak-Jankowska A, Appaix F, Louage B, Hamard L, et al. Thermoresponsive hyaluronic acid nanogels as hydrophobic drug carrier to macrophages. Acta Biomater. 2014; 10: 4750–4758. PubMed: https://pubmed.ncbi.nlm.nih.gov/25110287/
- Liang K, Ng S, Lee F, Lim J, Chung JE, et al. Targeted intracellular protein delivery based on hyaluronic acid-green tea catechin nanogels. Acta Biomater. 2016; 33: 142–152. PubMed: https://pubmed.ncbi.nlm.nih.gov/26785145/
- Water JJ, Kim Y, Maltesen MJ, Franzyk H, Foged C, et al. Hyaluronic acidbased nanogels produced by microfluidics-facilitated self-assembly improves the safety profile of the cationic host defense peptide novicidin. Pharm Res. 2015; 32: 2727–2735. PubMed: https://pubmed.ncbi.nlm.nih.gov/25813840/
- Park K, Lee MY, Kim KS, Hahn SK. Target specific tumor treatment by VEGF siRNA complexed with reducible polyethyleneimine-hyaluronic acid conjugate. Biomaterials. 2010; 31: 5258–5265. PubMed: https://pubmed.ncbi.nlm.nih.gov/20378167/
- Zhong Y, Zhang J, Cheng R, Deng C, Meng F, et al. Reversibly crosslinked hyaluronic acid nanoparticles for active targeting and intelligent delivery of doxorubicin to drug resistant CD44+ human breast tumor xenografts. J Control Release. 2015; 205: 144–154. PubMed: https://pubmed.ncbi.nlm.nih.gov/25596560/
- Oh EJ, Park K, Kim KS, Kim J, Yang JA, et al. Target specific and long-acting delivery of protein, peptide, and nucleotide therapeutics using hyaluronic acid derivatives. J Control Release. 2010; 141: 2–12. PubMed: https://pubmed.ncbi.nlm.nih.gov/19758573/
- Park JK, Shim JH, Kang KS, Yeom J, Jung HS, et al. Solid free-form fabrication of tissue-engineering scaffolds with a poly (lactic-co-glycolic acid) grafted hyaluronic acid conjugate encapsulating an intact bone morphogenetic protein-2/poly (ethylene glycol) complex. Adv Funct Mater. 2011; 21: 2906–2912.
- Fan Y, Deng C, Cheng R, Meng F, Zhong Z. In situ forming hydrogels via catalystfree and bioorthogonal “tetrazole-alkene” photo-click chemistry. Biomacromolecules. 2013; 14: 2814–2821.
- Song W, Wang Y, Qu J, Lin Q. Selective functionalization of a genetically encoded alkene-containing protein via “photoclick chemistry” in bacterial cells. J Am Chem Soc. 2008; 130: 9654–9655. PubMed: https://pubmed.ncbi.nlm.nih.gov/18593155/
- Ganesh S, Iyer AK, Gattacceca F, Morrissey DV, Amiji MM. In vivo biodistribution of siRNA and cisplatin administered using CD44-targeted hyaluronic acid nanoparticles. J Control Release. 2013; 172: 699–706. PubMed: https://pubmed.ncbi.nlm.nih.gov/24161254/
- Li J, Huo M, Wang J, Zhou J, Mohammad JM, et al. Redox-sensitive micelles self-assembled from amphiphilic hyaluronic aciddeoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials. 2012; 33: 2310–2320. PubMed: https://pubmed.ncbi.nlm.nih.gov/22166223/
- Yao HJ, Zhang YG, Sun L, Liu Y. The effect of hyaluronic acid functionalized carbon nanotubes loaded with salinomycin on gastric cancer stem cells. Biomaterials. 2014; 35: 9208–9223. PubMed: https://pubmed.ncbi.nlm.nih.gov/25115788/
- Kesharwani P, Banerjee S, Padhye S, Sarkar FH, Iyer AK. Hyaluronic acid engineered nanomicelles loaded with 3,4-difluorobenzylidene curcumin for targeted killing of CD44+ stem-like pancreatic cancer cells. Biomacromolecules. 2015; 16: 3042–3053. PubMed: https://pubmed.ncbi.nlm.nih.gov/26302089/
- Wang X, Zhang J, Cheng R, Meng F, Deng C, et al. Facile synthesis of reductively degradable biopolymers using cystamine diisocyanate as a coupling agent. Biomacromolecules. 2016; 17: 882–890.
- Yu Z, Ho LY, Lin Q. Rapid, photoactivatable turn-on fluorescent probes based on an intramolecular photoclick reaction. J Am Chem Soc. 2011; 133: 11912–11915. PubMed: https://pubmed.ncbi.nlm.nih.gov/21736329/
- Peng J, Qi T, Liao J, Fan M, Luo F, et al. Synthesis and characterization of novel dual-responsive nanogels and their application as drug delivery systems. Nanoscale. 2012; 4: 2694–2704.
- Shi Y, van Steenbergen MJ, Teunissen EA, Novo LS, Gradmann S, et al. π–π stacking increases the stability and loading capacity of thermosensitive polymeric micelles for chemotherapeutic drugs. Biomacromolecules. 2013; 14: 1826-1837. PubMed: https://pubmed.ncbi.nlm.nih.gov/23607866/
- Zhu Y, Wang XX, Chen J, Zhang J, Meng F, et al. Bioresponsive and fluorescent hyaluronic acid-iodixanol nanogels for targeted X-ray computed tomography imaging and chemotherapy of breast tumors. J Control Release. 2016; 244: 229–239. PubMed: https://pubmed.ncbi.nlm.nih.gov/27568289/
- Kobayashi H, Choyke PL. Target-cancer-cell-specific activatable fluorescence imaging probes: rational design and in vivo applications. Acc Chem Res. 2010; 44: 83–90. PubMed: https://pubmed.ncbi.nlm.nih.gov/21062101/
- Chauhan VP, Jain RK. Strategies for advancing cancer nanomedicine. Nat Mater. 2013; 12: 958–962. PubMed: https://pubmed.ncbi.nlm.nih.gov/24150413/
- Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010; 7: 653–664. PubMed: https://pubmed.ncbi.nlm.nih.gov/20838415/
- Liang L, Lin SW, Dai W, Lu JK, Yang TY, et al.Novel cathepsin B-sensitive paclitaxel conjugate: higher water solubility, better efficacy and lower toxicity. J Control Release. 2012; 160: 618–629. PubMed: https://pubmed.ncbi.nlm.nih.gov/22410114/
- Zou Y, Song Y, Yang W, Meng F, Liu H, et al. Galactose-installed photocrosslinked pH-sensitive degradable micelles for active targeting chemotherapy of hepatocellular carcinoma in mice. J Control Release. 2014; 193: 154–161. PubMed: https://pubmed.ncbi.nlm.nih.gov/24852094/
- Loutfy H. Madkour Book: Reactive Oxygen Species (ROS), Nanoparticles, and Endoplasmic Reticulum (ER) Stress-Induced Cell Death Mechanisms. Paperback. https://www.elsevier.com/books/reactive-oxygen-species-ros-nanoparticles-and-endoplasmic-reticulum-er-stress-induced-cell-death-mechanisms/madkour/978-0-12-822481-6
- Xu A, Chen J, Peng H, Han G, Cai H. Simultaneous interrogation of cancer omics to identify subtypes with significant clinical differences[J]. Front Genet. 2019; 10: 236. PubMed: https://pubmed.ncbi.nlm.nih.gov/30984238/
- Madkour LH. Book. Nanoparticles Induce Oxidative and Endoplasmic Reticulum Antioxidant Therapeutic Defenses. https://www.springer.com/gp/book/9783030372965?utm_campaign=3_pier05_buy_print&utm_content=en_08082017 &utm_medium=referral&utm_source=google_books#otherversion=9783030372972
- Chen J, Han G, Xu A, Cai H. Identification of Multidimensional Regulatory Modules through. Multi-graph Matching with Network Constraints. IEEE Trans Biomed Eng. 2020; 67: 987-998. PubMed: https://pubmed.ncbi.nlm.nih.gov/31295100/
- Madkour LH. Book: Nucleic Acids as Gene Anticancer Drug Delivery Therapy. 1st Edition. https://www.elsevier.com/books/nucleic-acids-as-gene-anticancer-drug-deliverytherapy/madkour/978-0-12-819777-6
- Madkour LH. Book: Nanoelectronic Materials: Fundamentals and Applications (Advanced Structured Materials) 1st ed. 2019. https://www.springer.com/gp/book/9783030216207
- Wang YXJ, Choi Y, Chen ZY, Laurent S, Gibbs SL. Molecular imaging: from bench to clinic. Biomed Res Int. 2014; 357258. PubMed: https://pubmed.ncbi.nlm.nih.gov/25610862/
- Wang J, Mi P, Lin G, Xiáng Y, Wáng J, et al. Imaging-guided delivery of RNAi for anticancer treatment. Adv Drug Deliv Rev. 2016; 104: 44–60. PubMed: https://pubmed.ncbi.nlm.nih.gov/26805788/